

Versión 2

Cuando Mendel descubrió las leyes que llevan su nombre demostró que la verdadera herencia que cada progenitor transmite a sus hijos es una dotación genética completa. Por su parte, Darwin cayó en la cuenta de que la conducta es un factor importante para la adaptación, medida por el éxito reproductivo; efectivamente, la conducta es variable y heredable, al menos hasta un cierto punto, como, por ejemplo, lo demuestra el éxito en la crianza selectiva de perros. Lo más probable es que la explicación de todas las diferencias entre perros y lobos sea de tipo comportamental, puesto que la diferencia esencial entre los perros y los demás cánidos tiene que ver con la domesticación. Al parecer los híbridos de lobos y perros son fértiles, lo que apunta a que no se ha completado el proceso de especiación. La teoría sintética de la evolución o neodarwinismo explica la evolución por selección natural como un cambio en las frecuencias alélicas de la población. Entonces, la cría selectiva que implicó la domesticación del lobo hasta convertirlo en perro, supuso un cambio en las frecuencias de determinados alelos, cambio genético que, supuestamente, explica las notables diferencias en la conducta de una y otra especie.
La conducta es un fenotipo bajo el cual subyace un genotipo que la explica (en mayor o menor medida dependiendo de cómo sea la interacción de esos genes con el ambiente). Igual que la diferencia de genotipos en los guisantes de Mendel explica las diferencias de color o textura de las semillas, una diferencia en los genotipos de lobos y perros debería explicar las diferencias entre ellos, tanto en apariencia física como en conducta. Hay una relación muy estrecha entre la genética molecular (estudio del material genético o ADN de los cromosomas y de los mecanismos bioquímicos que explican la expresión de la información contenida en ese ADN), genética mendeliana y teoría sintética de la evolución.
Mucho antes de que los descubrimientos de Mendel fueran reconocidos por la sociedad, Galton (1822-1911), basándose en los principios de la teoría de la evolución de Darwin, dedujo que todos los rasgos conductuales humanos debían tener una base genética, resultado de la selección natural, hipotetizó que la inteligencia humana tiene una base genética, y pretendió haberlo demostrado al comprobar que cuanto mayor era el grado de parentesco, mayor era la semejanza en eminencia intelectual. Por esto y por la enorme cantidad de investigaciones relacionadas que desarrolló o estimuló, se considera a Galton el fundador de la Genética de la Conducta.
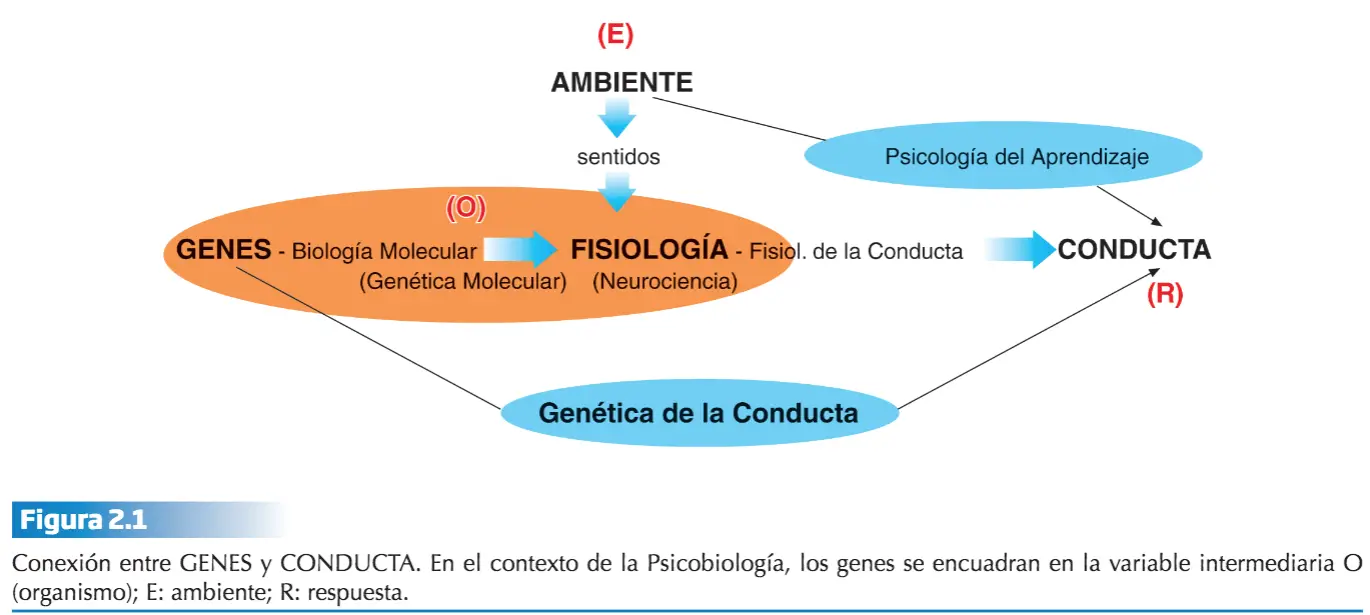
Estamos de acuerdo en que la conducta es un fenotipo, pero explicar la influencia de los genes sobre la conducta (el otro factor explicativo es el ambiente) exige rastrear todo el proceso, que va desde el ADN de los gametos que se unen para formar el cigoto (el nuevo ser vivo) hasta el rasgo fenotípico conductual cuya base genética queremos demostrar.
DESCUBRIMIENTO DE LA GENÉTICA: LAS LEYES DE MENDEL
«Una mayoría de autores consideran hoy que, en lo que se refiere a la explicación de transmisión de los caracteres, la denominada ley de la uniformidad no se debería considerar como tal ley, puesto que lo Mendel observó y se recoge como un enunciado de la primera ley, a saber que había uniformidad en los híbridos de plantas que él estudió, no es verdaderamente una explicación de una transmisión de caracteres, ya que la dominancia se relaciona con la expresión de los alelos y es un hecho que no tiene que ver con la transmisión. Por ello, después de 2009 en la nueva edición del manual de la asignatura, cuando ya se habían implantado los Grados en la Universidad española y hubo que preparar nuevos temarios, sólo se citaron como leyes la de Segregación (que es la primera) y la de Combinación independiente ( la segunda)». ED aLF
G. Mendel (1822-1884) hace publico su trabajo en 1866 «Experimentos en la hibridación de plantas», 35 años después su trabajo es reconocido en el mundo. Y no es que fuera desconocido el hecho de que los descendientes se parecen a sus progenitores y a veces incluso más a algún abuelo: el problema era que no había ninguna explicación material para dicha semejanza y además, parecía casi evidente que los descendientes presentan muy a menudo una apariencia intermedia de los rasgos de ambos progenitores, lo que parecía avalar la hipótesis de una herencia promedio. Mendel se convenció de que, puesto que los caracteres (hoy diríamos rasgos) de una raza establecida (raza pura) se mantienen constantes sólo si tanto el polen como el óvulo proceden de plantas con dichos caracteres, ambos gametos debían aportar algo a los rasgos de la planta: a ese algo Mendel lo llamó factor (pero en la actualidad lo llamamos gen). Gracias a sus experimentos con polinización artificial cruzada entre variedades puras de guisantes (Pisum sativum) demostró que dichos factores no se mezclan, sino que se conservan íntegros como unidades de trasmisión hereditaria.
Mendel, debe su fama (póstuma) y su éxito en el campo de lo que luego se conocería como Genética, no sólo a su meticulosidad experimental sino también a la toma de decisiones a la hora de diseñar y analizar
sus experimentos: su hipótesis le llevó a fijarse en rasgos discretos o cualitativos, para lo que eligió como material de estudio diversas variedades puras de plantas de guisantes en las que se podían discernir con
claridad rasgos dicotómicos. Dedujo que trataba de variedades (razas) puras por el hecho de que durante 8 generaciones la autopolinización de cada raza dio siempre descendientes idénticos (fenotípicamente) a sus progenitores. De hecho, estas razas puras le sirvieron de grupo control respecto a los grupos de plantas híbridas, que le permitieron demostrar las posteriormente llamadas Leyes de Mendel.
- LEY DE LA UNIFORMIDAD. Aunque el manual de estudio no le atribuye esta ley a Mendel, de hecho en texto del capítulo no aparece la palabra «uniformidad», sin embardo el el video siguen otro criterio. Esta Ley establece que si se cruzan dos líneas puras para un determinado carácter, los descendientes de la primera generación serán todos iguales entre sí, fenotípica y genotípicamente, e iguales fenotípicamente a uno de los progenitores (de genotipo dominante), independientemente de la dirección del cruzamiento. Expresado con letras mayúsculas las dominantes y minúsculas las recesivas.
- LEY DE LA SEGREGACIÓN. Esta ley establece que durante la formación de los gametos, cada alelo de un par se separa del otro miembro para determinar la constitución genética del gameto filial. Es muy habitual representar las posibilidades de hibridación mediante un cuadro de Punnett.
- LEY DE LA COMBINACIÓN INDEPENDIENTE. En ocasiones es descrita como la 2.ª ley, en caso de considerar solo dos leyes (criterio basado en que Mendel solo estudió la transmisión de factores hereditarios y no su dominancia/expresividad). Mendel concluyó que diferentes rasgos son heredados independientemente unos de otros, no existe relación entre ellos, por lo tanto el patrón de herencia de un rasgo no afectará al patrón de herencia de otro. Solo se cumple en aquellos genes que no están ligados (es decir, que están en diferentes cromosomas) o que están en regiones muy separadas del mismo cromosoma. En este caso la descendencia sigue las proporciones. Representándolo con letras, de padres con dos características AALL y aall (donde cada letra representa una característica y la dominancia por la mayúscula o minúscula), por entrecruzamiento de razas puras (1.ª Ley), aplicada a dos rasgos, resultarían los siguientes gametos: AL x al = AL, Al, aL, al.
Establece que si se cruzan dos líneas puras para un determinado carácter, los descendientes de la primera generación serán todos iguales entre sí, fenotípica y genotípicamente, e iguales fenotípicamente a uno de los progenitores (de genotipo dominante), independientemente de la dirección del cruzamiento. Cuando se cruzan dos individuos de raza pura (homocigotos), la primera generación filial (heterocigotos), será igual entre ellos (fenotipos y genotipos) y, además, sobresaldrá el rasgo fenotípico de uno de los progenitores (genotipo dominante).

Un cruce monohíbrido, en genética, hace referencia al cruce de dos individuos que difieren en un solo carácter o rasgo. En términos más exactos, los individuos poseen dos variaciones o “alelos” de la característica a ser estudiada. Los resultados de la primera generación de un cruce monohíbrido aportan la información necesaria para inferir el genotipo de los organismos parentales.
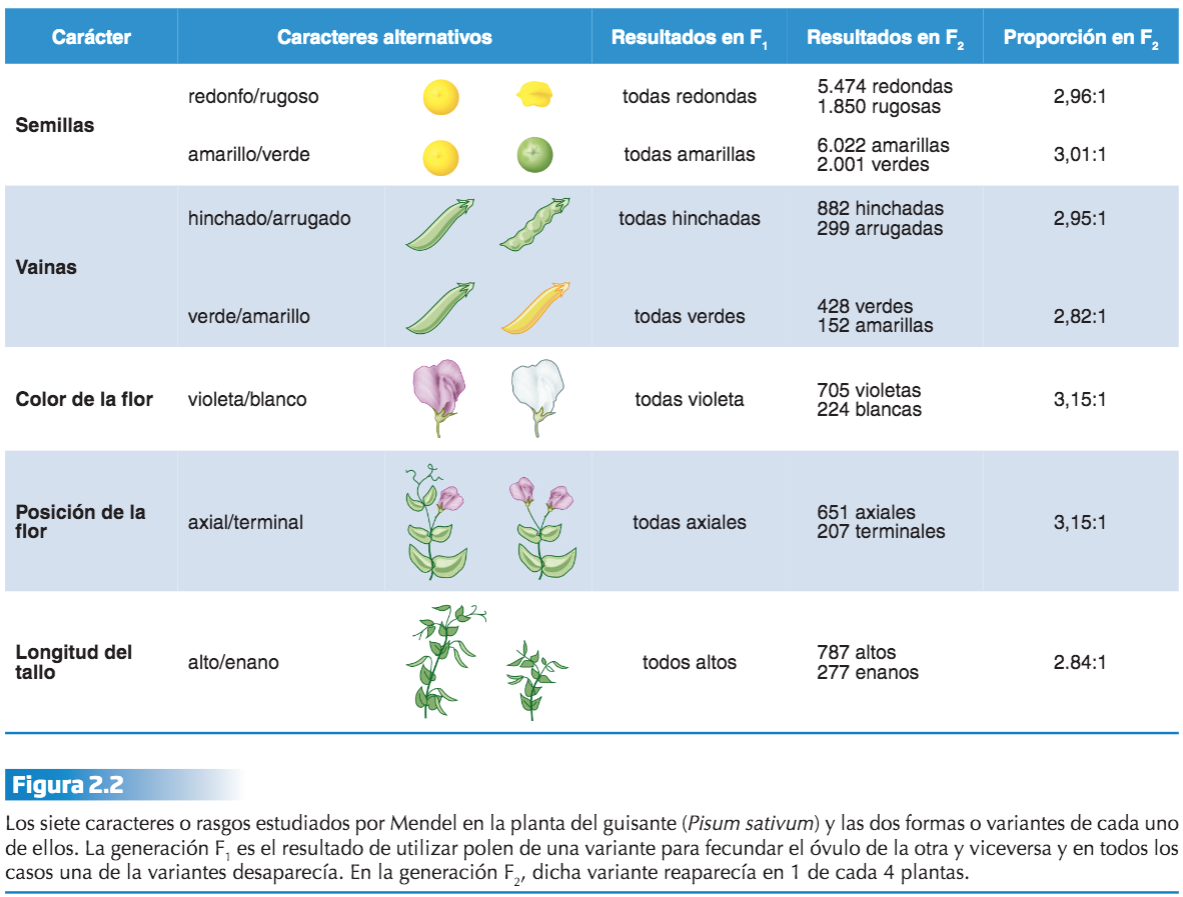
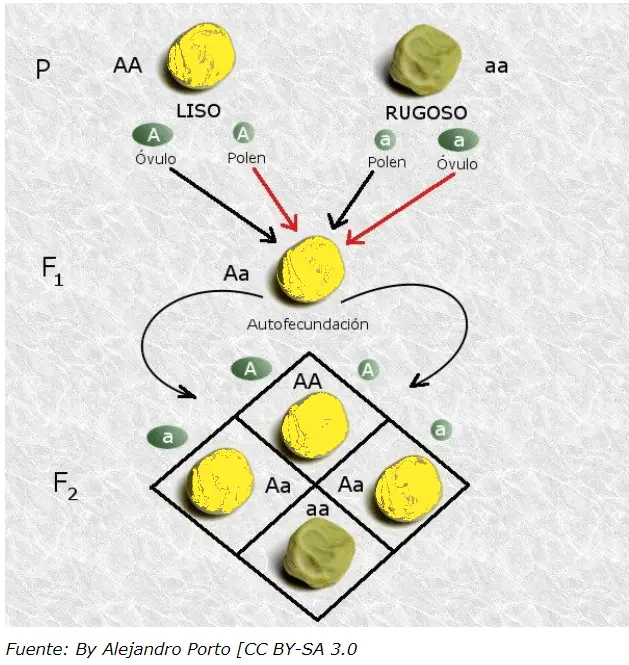
Mendel estudio rasgos o caracteres; cruzo entre sí dos razas puras que diferían en uno de los rasgos concretos (generación parental P), p. ej., plantas de semilla lisa con plantas de semilla rugosa; plantas altas con plantas enanas, etc. Así comprobó que todos los descendientes de estos cruces (generación F1 ) presentaban una apariencia en el rasgo considerado, idéntica a uno de los dos progenitores, independientemente además de si era el que aportó el óvulo o el polen. A la forma del carácter que se manifiesta (fenotipo) en esta generación F1, Mendel lo califico como dominante. Del cruce de plantas altas con plantas enanas resultó una planta híbrida donde el 100% fueron altas (tan altas al menos como las plantas de la raza alta utilizada en la fecundación); y lo mismo ocurrió con todos los rasgos dominantes estudiados. A la variante del carácter que no aparece en esta generación F1, la llamó recesiva. A juicio de Mendel, para estos 7 caracteres, no se produce el efecto de mezcla hereditaria que cabía esperar.
Ley de la Segregación
Mendel estudió la herencia de rasgos en plantas de guisantes. Propuso un modelo donde pares de «elementos heredables», o genes, especifican rasgos. En sus experiemntos con las flores de los guisantes, del total de descendientes que obtuvo, 705 presentaban la flor violeta y 224 la flor blanca, 3/4 de la generación F2 resultante de la autofecundación natural de las plantas de la F1 presentaban el fenotipo dominante, mientras que el 1/4 restante manifestaba el fenotipo recesivo, es decir, el fenotipo que no aparecía en la generación F1. Mendel utiliza las matemáticas para concluir que la clave estaba en la proporción 3:1 en cada uno de los fenotipos. El hecho de que la forma recesiva del carácter reapareciese en la F2, Mendel lo interpreta como una consecuencia de que ésta no había desaparecido en la F1: simplemente, por alguna razón, no se manifestaba en la F1 . Esta variante, recesiva, quedaba oculta. Para Mendel estos resultados indicaban que cada carácter (color de la flor, aspecto de la semilla, etc) era debido a un elemento o factor hereditario que corresponde al término moderno de gen.
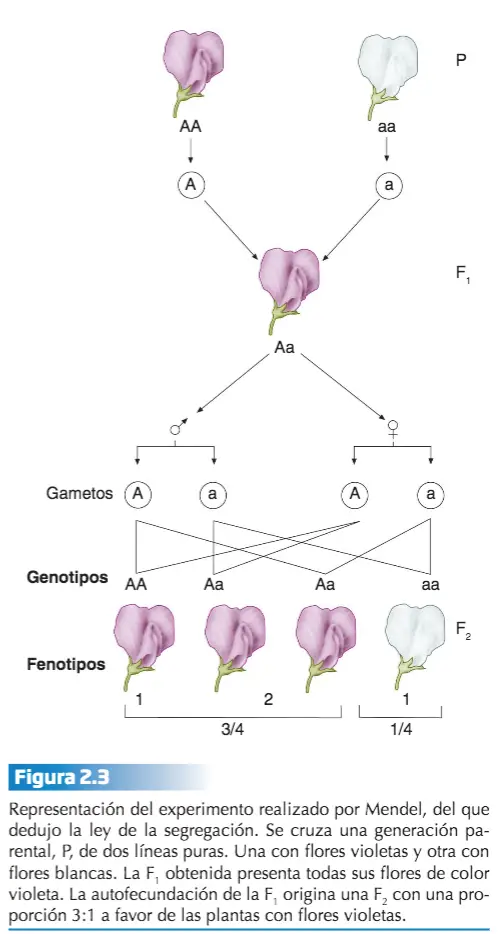
Los genes vienen en diferentes versiones o alelos. Un alelo dominante esconde al alelo recesivo y determina la apariencia del organismo. Cuando un organismo hace gametos, cada gameto recibe solo una copia del gen, que es seleccionada al azar. Esto se conoce como la ley de la segregación. En el caso del gen para el carácter color de la flor, existe en dos formas o variantes, la responsable del color violeta y la causante de que la flor sea blanca. A estos genes que presentan más de una variante se les llama alelomorfos o, simplemente, alelos. Cada planta porta dos genes para cada carácter, uno procedente de la planta materna y otro de la paterna o, cuando hay autofecundación, del gameto femenino y del gameto masculino, respectivamente. En el caso que nos ocupa, el gen responsable del color de la flor es alelomorfo, presenta dos alelos, que podemos representar por la letra A, para el alelo dominante, y la letra a, para el recesivo.
Durante la formación de los gametos (gracias al proceso de meiosis) los alelos se separan (segregan), de forma que cada gameto recibe un solo alelo. Al juntarse dos gametos se restablece en el nuevo individuo la dotación doble habitual para cada carácter. La tabla de Punnett puede utilizarse para predecir genotipos (combinaciones de alelos) y fenotipos (rasgos observables) de la descendencia de cruces genéticos.
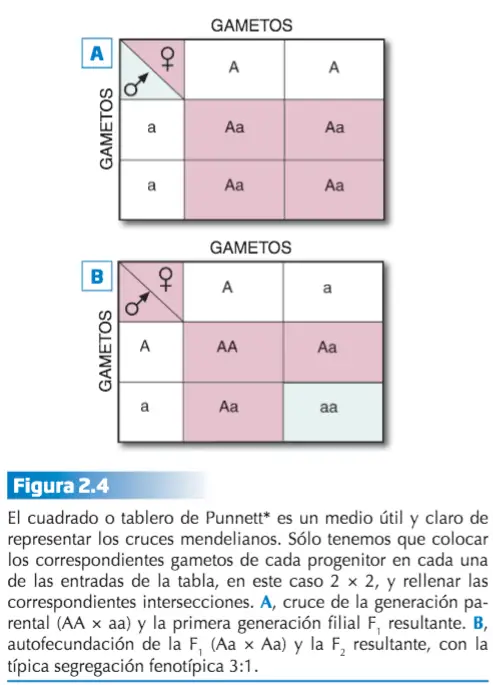
La constitución genética en relación a un carácter, o a todos los caracteres, se denomina genotipo y la manifestación externa del genotipo, fenotipo. P. ej., el genotipo de un híbrido es Aa y su fenotipo, el color violeta. Por su parte, los genotipos pueden ser de dos tipos: homocigotos, si los dos alelos son iguales, AA o aa, y heterocigotos, cuando los dos alelos son diferentes, Aa. Por tanto, los homocigotos sólo podrán producir un tipo de gameto según el alelo que portan, mientras que los heterocigotos producirán dos tipos, unos con el alelo A y otros con el alelo a.
Basicamente así es como Mendel extrajo la ley de la segregación: las variantes recesivas enmascaradas en la F1 heterocigota, resultante del cruce entre dos líneas puras (homocigotas, por tanto), reaparecen en la segunda generación filial en una proporción 3:1, debido a que los miembros de la pareja de alelos del heterocigoto se separan sin experimentar alteración alguna durante la formación de los gametos. Dado que, fenotípicamente hablando, los homocigotos dominantes y los heterocigotos son indistinguibles, una manera de averiguar a qué genotipo corresponde un determinado fenotipo es a través del denominado cruzamiento prueba.
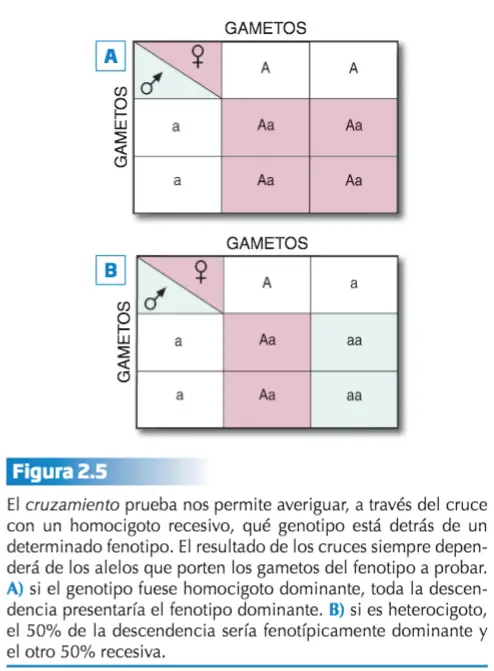
Ley de la Combinación Independiente
Mendel estudió la herencia simultánea de dos caracteres diferentes (dos loci situados sobre cromosomas diferentes), tales como el color de la semilla, que tiene dos variantes, amarilla o verde, y el aspecto de ésta, cuyas variantes son lisa y rugosa. Para ello cruzó dos líneas puras, una de plantas con semillas amarillas y lisas, y otra cuyas semillas eran verdes y rugosas.
La autofecundación de las plantas de la F1 proporcionó una generación F2 constituida por las cuatro combinaciones posibles para los caracteres estudiados: semillas amarillas y lisas, amarillas y rugosas, verdes y lisas, y verdes y rugosas, con unas proporciones respectivas de 9:3:3:1. Considerados de forma independiente, cada carácter seguía presentándose en una proporción 3:1.
Mendel hizo uso del cálculo de probabilidades: sabiendo que la probabilidad del fenotipo dominante es 3/4, la probabilidad de que se den conjuntamente ambos rasgos dominantes (amarillo y liso) es 3/4 x 3/4, y sucede lo mismo con los rasgos recesivos. En el caso de la combinación de un rasgo dominante con otro recesivo (amarillo con rugoso, o verde con liso), la probabilidad, igualmente, es 3/4 x 1/4 y 1/4 x 3/4 respectivamente. Los valores que estas probabilidades nos dan corresponden exactamente al patrón 9:3:3:1.Nótese la equivalencia (1/4 x 1/4): 1:16, así como la equivalencia (3/4 x 3/4): 9:1 6, etc.
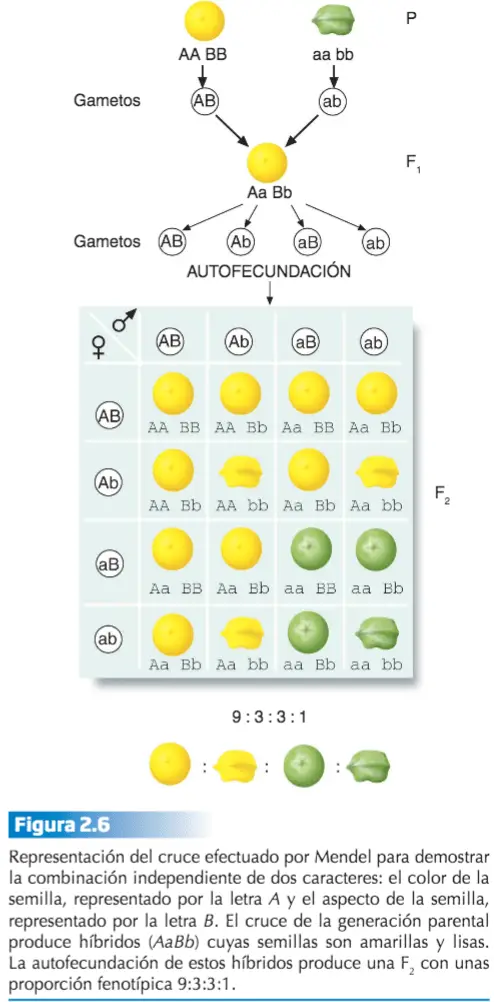
De estos resultados Mendel dedujo la ley de la combinación independiente: los miembros de parejas alélicas diferentes se segregan o combinan independientemente unos de otros cuando se forman los gametos.
LA REPRODUCCIÓN SEXUAL Y LAS LEYES DE MENDEL: MEIOSIS Y TEORÍA CROMOSÓMICA DE LA HERENCIA
La genial intuición mendeliana de que los progenitores pasan a sus descendientes una copia de cada factor hereditario (gen) de las dos que poseen, es un hecho comprobado:
Meiosis es el nombre que recibe el proceso por el que se reparten equitativamente y al azar los genes entre los gametos, de forma que cada gameto recibe una sola copia de cada gen.
- Los gametos han de recibir una copia, y sólo una, de cada cromosoma, de forma que cada gameto reciba una copia de cada gen, de las dos que normalmente poseen las células de los organismos pluricelulares.
- Aquí los conceptos esenciales son diploide y haploide. Las células somáticas normales son diploides, ya que portan dos copias de cada cromosoma (y por ende, de cada gen). Podemos decir que los cromosomas van por parejas en el sentido de que contienen los mismos genes: cada cromosoma de cada una de esas parejas tiene su homólogo. El número diploide de cromosomas se representa con la expresión 2n. Puesto que los gametos son haploides porque sólo contienen una copia de cada cromosoma, su número se representa con n. Los gametos de los guisantes de Mendel, por ejemplo, poseen 7 cromosomas (n=7) y sus células somáticas 14 cromosomas (2n); la especie humana es también diploide y el número normal de cromosomas de nuestras células es de 46 (2n=46); los óvulos y espermatozoides humanos, por su parte, contienen sólo 23 cromosomas (n=23) .
El desarrollo de cualquier organismo vivo pluricelular implica reproducción celular: mediante mitosis, las células se multiplican de forma que una célula se divide y da lugar a dos células idénticas: esto implica que las células somáticas humanas que se dividen han de hacer previamente dos copias exactas de todo su material genético y luego, distribuirlo exactamente entre las dos células resultantes de la mitosis. Esto significa que han de sintetizar una copia nueva de cada cromosoma, de forma que cada una de las dos células hijas resultantes tenga a su vez 2n cromosomas, o sea, 46 cromosomas.
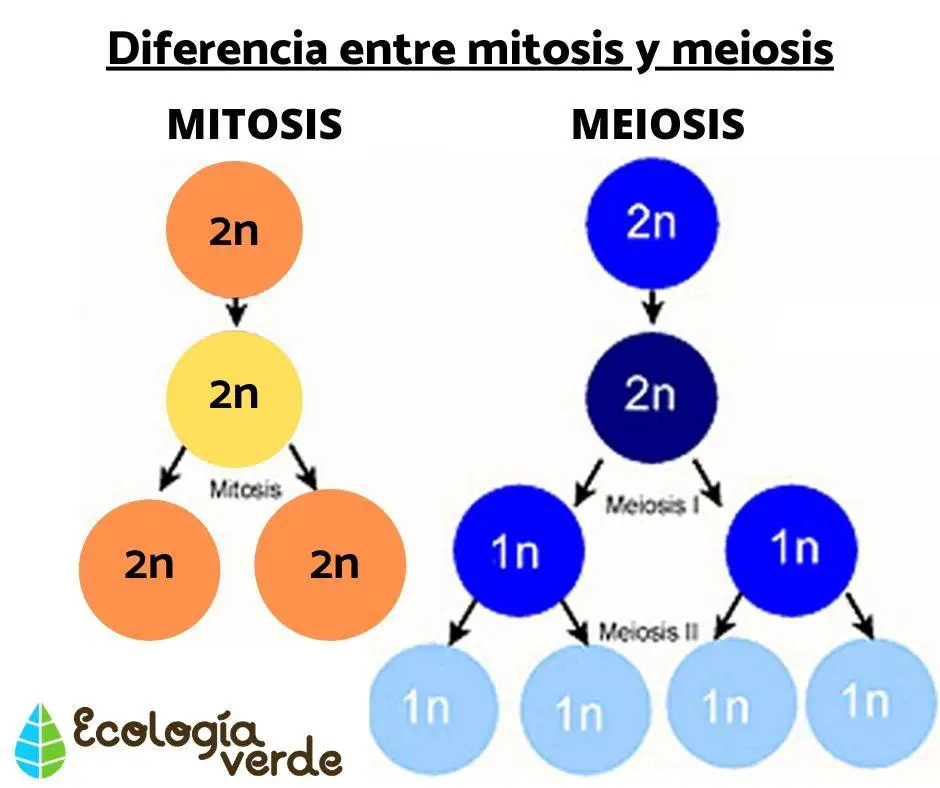
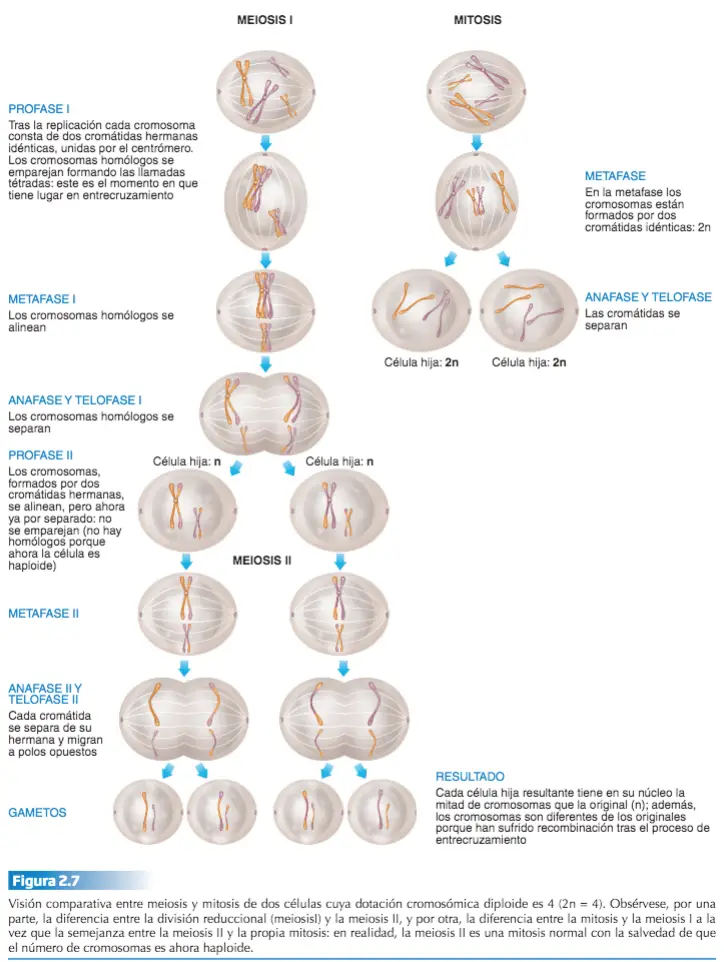
Tanto en la mitosis como en la meiosis I, al menos hasta la metafase, los cromosomas están formados por dos cromátidas, puesto que la molécula de ADN original ha de haberse replicado, de forma que en este momento del proceso, las células tienen un número diploide de cromosomas formados cada uno por dos cromátidas.
Diferencia entre mitosis y meiosis I
- Mitosis. Al dividirse en dos la célula original, las células hijas reciben cada una una cromátida de cada cromosoma (con lo que se mantiene el número diploide, sólo que ahora se habla de cromátidas como sinónimo de cromosoma),
- Meiosis I. Las dos células resultantes reciben cada una un número haploide de cromosomas (la mitad de los cromosomas), pero todavía constituidos por dos cromátidas cada uno: si en la mitosis humana las dos células resultantes de la división celular contienen 46 cromosomas formados por una sola cromátida cada uno, en el caso de la meiosis I, el resultado son dos células con 23 cromosomas cada una, pero en este caso formados por dos cromátidas.
La Meiosis Propiamente Dicha
La reproducción de los organismos pluricelulares parte de una única célula original, llamada zigoto, y que por divisiones sucesivas (mitosis) y diversos procesos de especialización, terminan dando lugar a otros organismos complejos. En la reproducción sexual cada progenitor aporta una sola copia de cada gen, y es preciso un mecanismo que explique cómo es posible que se formen gametos (que al unirse en la fecundación forman el zigoto) con una sola copia de todos y cada uno de los genes. Lo esencial para nosotros es entender el proceso por el cual se logra formar gametos haploides a partir de precursores diploides.
La meiosis se lleva a cabo en dos etapas:
- Meiosis l. Consiste en dividir la célula (2n) de tal forma que cada célula hija reciba un único y completo juego de cromosomas de la célula madre, es decir, pase a ser haploide (n) . Cada célula hija recibe un miembro de cada una de las parejas de cromosomas, sólo uno de los cromosomas homólogos.
- Meiosis II. Consiste en una división normal, equivalente a una mitosis, de las células obtenidas en la primera etapa.
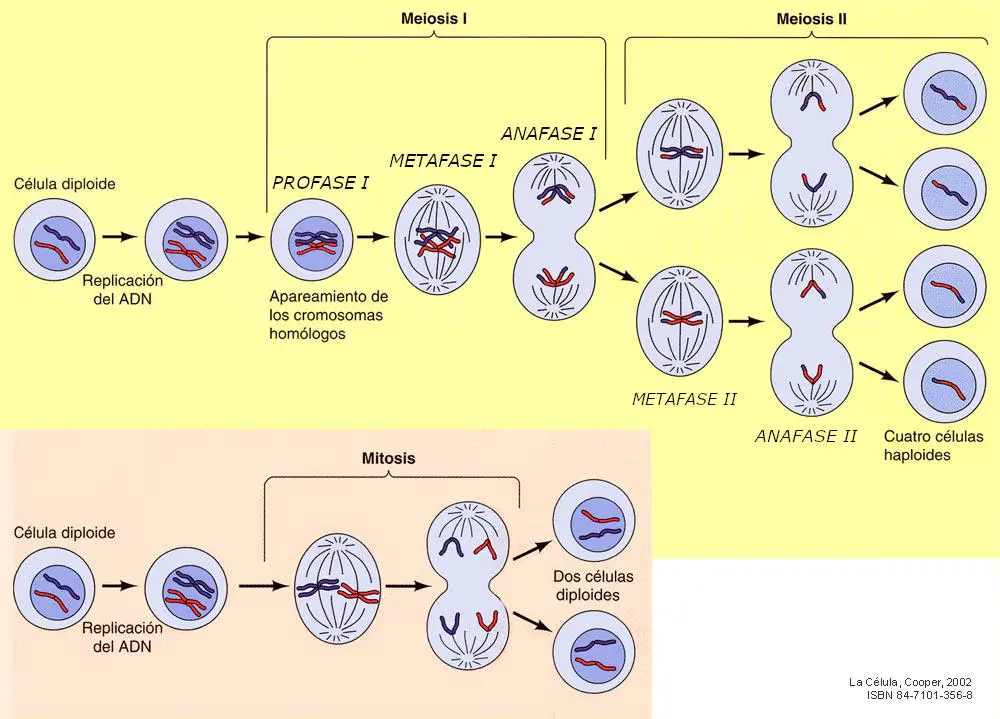
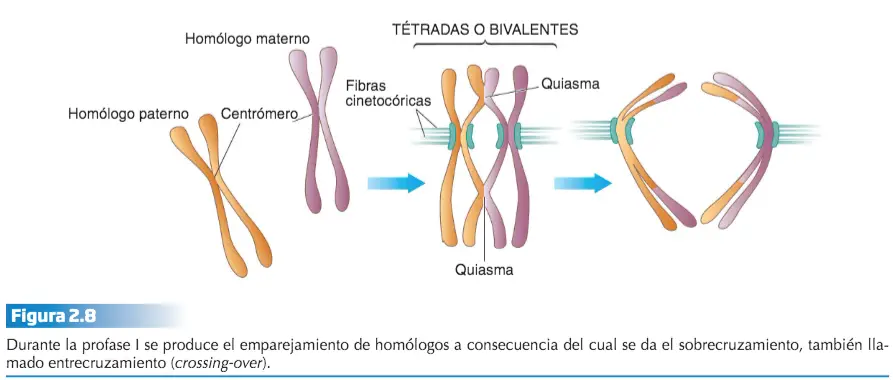
La meiosis I comienza con la profase l. Ésta difiere de la profase mitótica en que los cromosomas homólogos se aparean dos a dos, punto por punto, a lo largo de toda su longitud, formando lo que se denominan bivalentes o tétrada, en referencia a las cuatro cromátidas del bivalente. El apareamiento de los homólogos tiene una importancia extraordinaria. A través de él se produce el fenómeno citológico del entrecruzamiento, mediante el cual se lleva a cabo la recombinación génica, el intercambio de genes de un cromosoma homólogo a otro.
La siguiente etapa es la metafase l. En ella los bivalentes o tétradas, mediante sus centrómeros, se insertan en las fibras del huso adoptando una ordenación circular sobre la placa ecuatorial. La metafase I continúa con la anafase I en la que, a diferencia de la anafase de una mitosis normal en la que se separan 2n cromátidas, en ésta se separan los cromosomas de los bivalentes, emigrando n cromosomas (cada uno con sus dos cromátidas) a cada polo. Finalmente, en la telofase I los cromosomas se sitúan en ambos polos de la célula, se desespiralizan y se produce la citocinesis, dando lugar a dos células hijas con n cromosomas. Por haberse reducido el número de cromosomas a la mitad, a esta división meiótica se le denomina también división reduccional.
Como se ha indicado, los cromosomas no han seguido durante la meiosis I el mismo comportamiento
que muestran durante la mitosis, ya que aquí se han separado cromosomas homólogos y no cromátidas.
Este movimiento cromosómico es la demostración citológica de la ley de la segregación.
La meiosis II es prácticamente igual que la mitosis, salvo por el hecho de que la célula que entra en división es haploide, ya no hay cromosomas homólogos y, por tanto, tras ella se obtienen dos células hijas con n cromátidas.
Recombinación y Ligamiento
En la profase I se efectúa el emparejamiento de los cromosomas homólogos dos a dos formando las llamadas tétradas o bivalentes.
Durante el emparejamiento de los cromosomas homólogos se producen intercambios de alelos entre los cromosomas de la pareja de homólogos. Este suceso se denomina sobrecruzamiento o entrecruzamiento y se pone de manifiesto citológicamente por la aparición, entre las cromátidas de los bivalentes, de puntos de cruce, en forma de «X», que se denominan quiasmas.
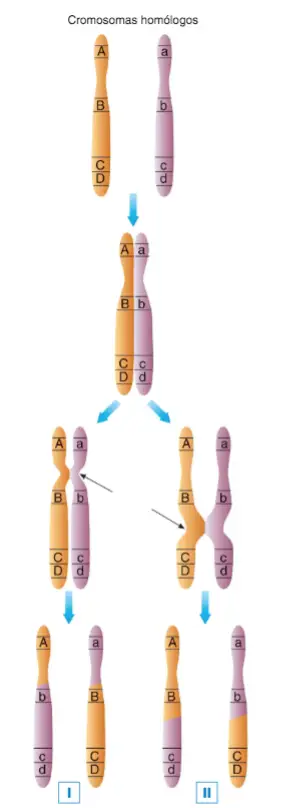
Durante la profase I de la meiosis, el sobrecruzamiento provoca
la recombinación génica, proceso mediante el cual los cromosomas homólogos intercambian trozos de ADN que contienen alelos concretos, de forma que los cromosomas resultantes tienen una combinación nueva de alelos: de esta forma, los cromosomas que forman parte de los gametos tienen combinaciones de alelos diferentes de las que tenían los cromosomas de los progenitores; podemos decir que gracias a la recombinación, cada cromosoma que un progenitor pasa a su descendiente es una combinación de los dos cromosomas homólogos que dicho progenitor recibió de su padre y de su madre. La recombinación génica es tanto más probable cuanto más alejados entre sí se hallan dos genes distintos, puesto que hay mucho espacio entre ellos para que se pueda formar un quiasma. El locus A está bastante separado del locus B (I) y de los loci C D (II), de forma que es bastante probable que ocurra un quiasma que permita su recombinación. Sin embargo, la distancia entre los loci C y D es tan pequeña, que es muy improbable que el sobrecruzamiento se dé entre ellos, ya sea en el caso de que el quiasma se produzca como en la secuencia I o como en la II (cuando se produce el sobrecruzamiento los cromosomas presentan dos cromátidas, sin embargo, para mayor claridad, en este dibujo sólo se representa una cromátida de cada cromosoma homólogo).
El porcentaje de recombinación entre dos loci está directamente relacionado con la distancia física que los separa dentro del cromosoma. A más distancia, más porcentaje de recombinación, y viceversa.
La consecuencia de la recombinación génica es la aparición, en un mismo cromosoma del gameto, de alelos que o bien proceden del padre, o bien de la madre de quien ha producido el gameto. El número de gametos distintos que se pueden formar mediante este proceso está en función de cuantos loci heterocigotos existen en un individuo, y esa cantidad se obtiene elevando el número 2 (par de homólogos) a la cifra de esos loci heterocigotos. La gran importancia de la recombinación génica es la variabilidad que genera. En nuestra especie se estima que en cada persona existen unos 3.350 loci en heterocigosis. Esto quiere decir que cada individuo puede formar 23350 gametos distintos, un número superior al de átomos existentes en el universo.
No siempre es posible efectuar intercambios entre los loci de los cromosomas homólogos mediante el sobrecruzamiento, cuanto más juntos estén dos loci, menos probabilidad habrá de que exista sobrecruzamiento entre ellos por un impedimento meramente físico. Cuando dos genes tienen nula o muy baja tasa de recombinación entre ellos, se dice que existe ligamiento entre esos dos genes o, simplemente, que están ligados. Si ocurre esto, no existe combinación independiente de caracteres y, por tanto, la ley de la combinación independiente de Mendel queda enmascarada.
DÓNDE ESTÁN Y QUÉ SON LOS GENES: EL CROMOSOMA EUCARIÓTICO Y LA NATURALEZA DEL MATERIAL HEREDITARIO
Los genes son las unidades de almacenamiento de información genética, segmentos de ADN que contienen la información sobre cómo deben funcionar las células del organismo. Tienen elementos que indican de dónde a dónde se tiene que leer, y su contenido determina la composición de las proteínas que se forman. Los genes se encuentran en unas estructuras diminutas parecidas a los espaguetis, que reciben el nombre de «cromosomas». Y los cromosomas se encuentran en el interior de nuestras las células.
Los cromosomas eucarióticos son moléculas muy largas de ADN doble hélice en interacción con proteínas (histonas y no histonas) que se pueden encontrar desde estados relajados o poco compactados como en los núcleos de las células en interfase hasta en estados altamente compactados como sucede en la metafase mitótica.
El descubrimiento del ligamiento y la recombinación propios de la meiosis permitió a principios del siglo xx poner de manifiesto que los genes se encontraban en los cromosomas de una forma ordenada. Es decir, que un determinado gen tiene una posición fija y concreta dentro del cromosoma.
En el caso de la especie humana, cada célula diploide posee 46 cromosomas; además, sabemos que al tratarse de una especie diploide, hay dos copias de cada cromosoma, por lo que en realidad sólo hay 23 cromosomas diferentes (24 en el caso de los varones), puesto que en los mamíferos el sexo está determinada por la presencia en de un cromosoma especial, el cromosoma Y.
Estructura del Cromosoma Eucariótico
Desde el punto de vista de la genética, un cromosoma es una molécula gigantesca de Ácido Desoxirribonucleico (ADN). El ADN es la mayor de las moléculas que portan los seres vivos. La longitud de todo el ADN que se encuentra en el total de las células del cuerpo humano es de alrededor de 2 x 1011 km. Cada cromosoma está constituido por una sola molécula de ácido desoxirribonucleico (ADN) unido a proteínas.
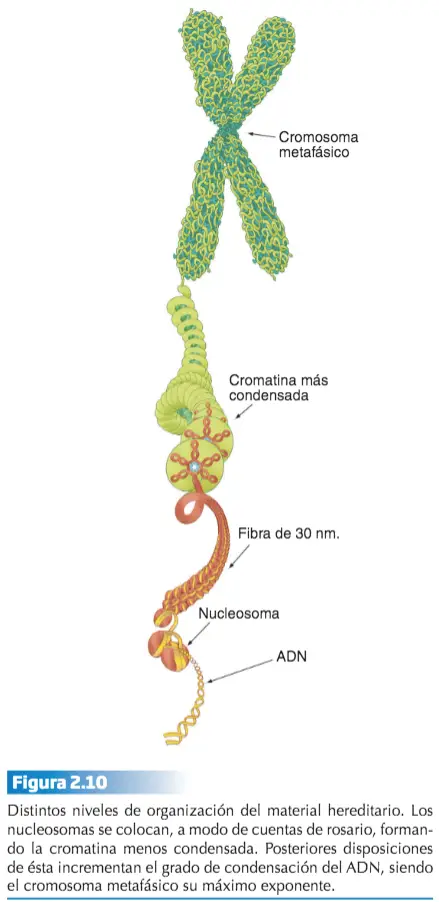
En las eucariotas, el aspecto del material hereditario varía desde la estructura claramente definida que representa el cromosoma metafásico, a una estructura amorfa y disgregada durante la interfase celular, que recibe el nombre de cromatina. El nivel de organización más elemental (dejando a un lado el que representa la propia molécula de ADN) es el que se alcanza a través de la unión de varios tipos de histonas con el ADN. Esta unión da lugar a una estructura denominada nucleosoma que representa la unidad básica de condensación del ADN.
Estas diferencias en el grado de condensación están directamente relacionadas con la funcionalidad del ADN: para que la información contenida en el ADN, lo que llamamos genes, se exprese en la célula es preciso que la molécula sea accesible, por lo que ha de estar poco o nada condensada, mientras que cuando hace falta traspasar esa misma información genética a las células hijas (mitosis) o a los gametos (meiosis), el ADN ha de estar empaquetado, muy condensado.
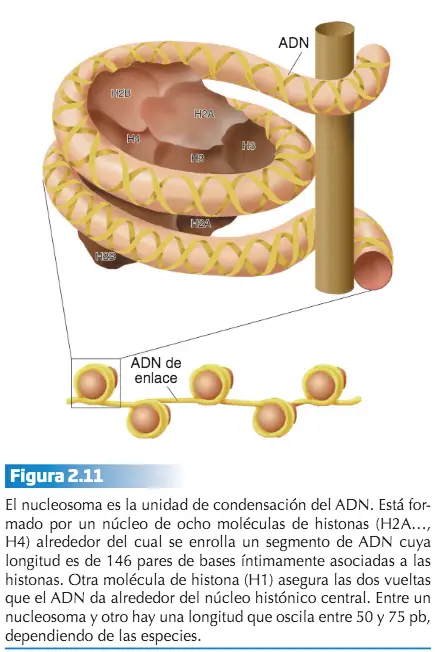
En los eucariotas, el estado menos condensado del ADN consiste en una estructura formada por nucleosomas distribuidos más o menos periódicamente a lo largo del material hereditario. Esta disposición hace que el ADN disminuya aproximadamente siete veces su longitud.
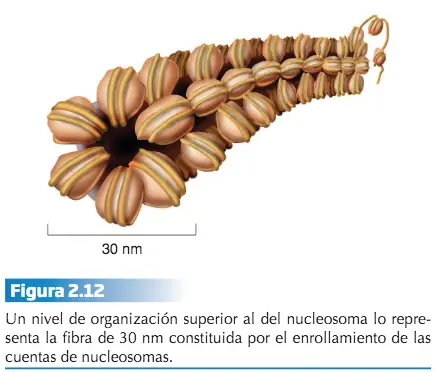
El nucleosoma representa el primer nivel y el cromosoma metafásico el último. Los sucesivos niveles se consiguen gracias a que los nucleosomas, una vez formados, se pliegan unos sobre otros de una manera ordenada formando una fibra de 30 nm de espesor. Esta fibra representa el siguiente nivel de organización del material hereditario y proporciona una compactación del ADN. Los siguientes niveles de condensación no son bien conocidos. La condensación del ADN a lo largo del ciclo celular varía desde el estado de cromatina al de cromosoma metafásico. Sin embargo, la cromatina tampoco presenta un estado homogéneo de compactación y se distinguen, a este respecto, dos tipos de cromatina:
- Eucromatina, presenta un empaquetamiento menor.
- Heterocromatina, que es la porción de cromatina más condensada.
Naturaleza Química del Material Hereditario
¿Cuál es la naturaleza química del gen? Las propiedades que debía cumplir el material encargado de portar la herencia biológica eran principalmente dos: guardar información y permitir la copia veraz de dicha información. Además, sus propiedades tenían que explicar la cierta capacidad de cambio o de alteración de la propia materia hereditaria, que daría cuenta de la existencia de variantes genéticas o alelos.
1869. F. Miescher (1844-1895), consigue aislar por primera vez el ácido desoxirribonucleico o ADN.
1953. J.D. Watson (1928-) y F. Crick (1916-2004) describen por primera vez la estructura de la molécula de ADN.
Estos descubrimientos contribuyeron definitivamente al desarrollo de la Genética molecular, una disciplina entre cuyos cometidos se encuentra establecer de una forma completa la relación entre genotipo y fenotipo. Igual que otros ácidos nucleicos, el ADN es una larga cadena doble formada por nucleótidos que son sustancias compuestas por una molécula de ácido fosfórico, una de un hidrato de carbono (ribosa o desoxirribosa) y otra de una base nitrogenada (púrica o pirimidínica). En el ADN estos nucleótidos forman dos cadenas, cada una de las cuales está dispuesta en espiral, enroscada una sobre otra formando una doble hélice.
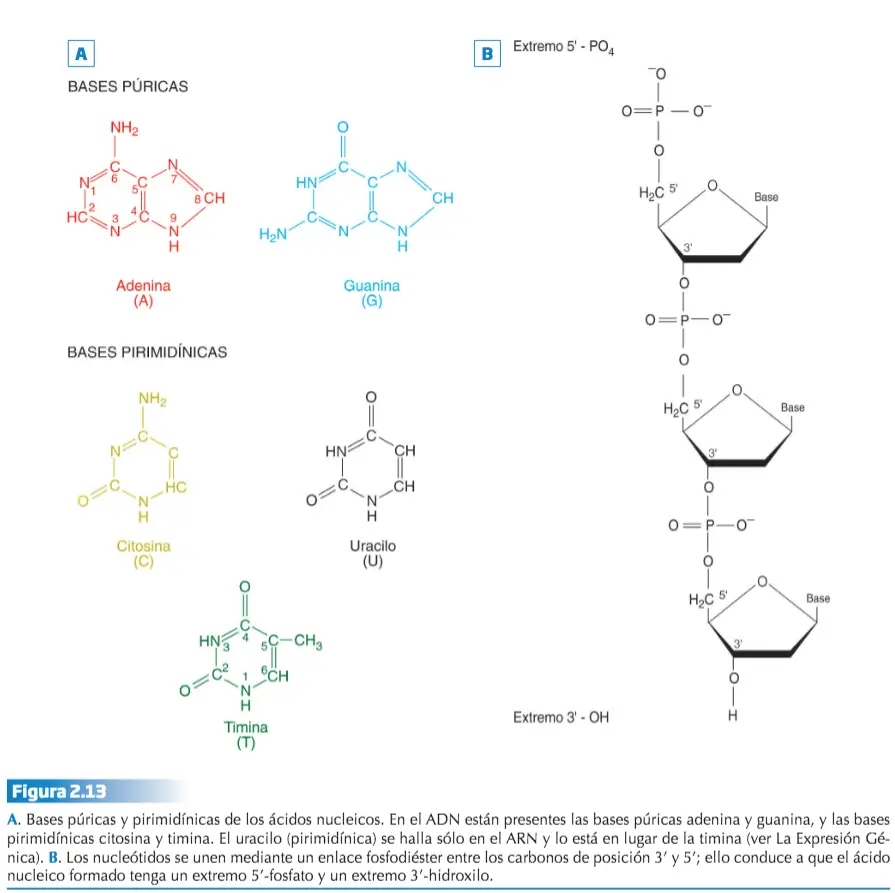
La doble hélice se consigue gracias a una disposición concreta de las moléculas que forman cada nucleótido del ADN. La espiral la marca la sucesión de las moléculas de desoxirribosa y ácido fosfórico de cada nucleótido, mientras que las bases nitrogenadas se sitúan en el interior. La unión entre las dos cadenas de nucleótidos que forman el ADN se lleva a cabo a través de puentes de hidrógeno que se establecen entre las bases púricas de una cadena y las pirimidínicas de la otra.
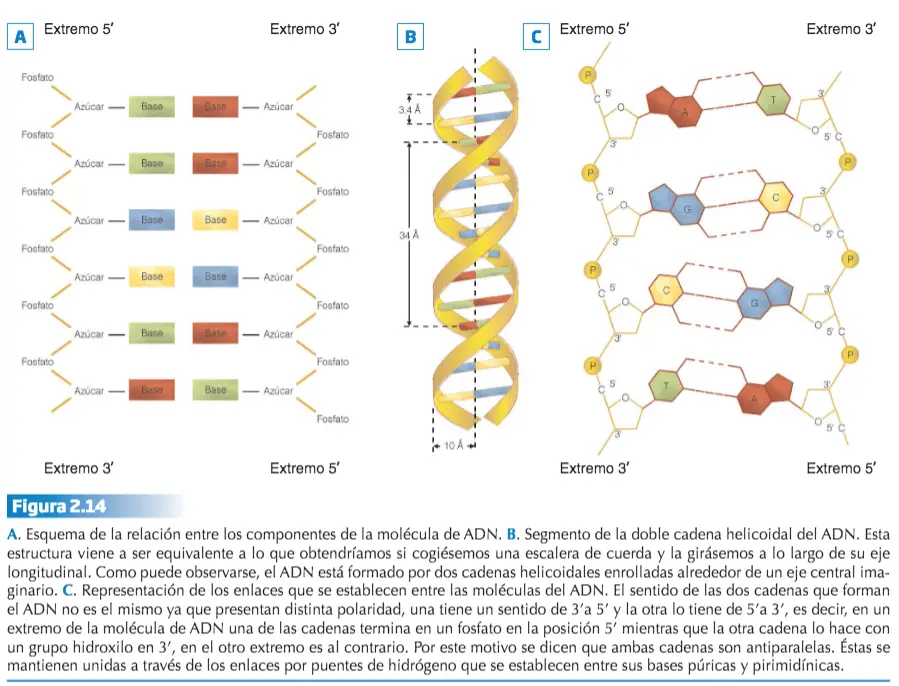
Existen severas restricciones acerca de las uniones entre las bases nitrogenadas de las dos hebras o cadenas que constituyen el ADN, ya que la adenina se aparea únicamente con la timina, mientras que la citosina lo hace sólo con la guanina. A esta relación restrictiva entre las bases se le denomina complementariedad.
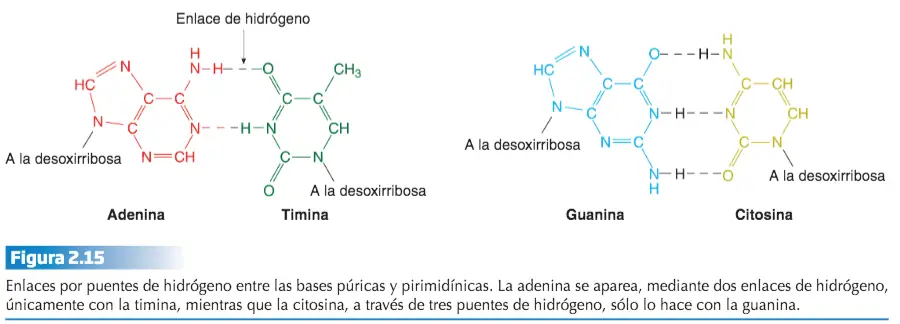
El modelo propuesto por Watson y Crick explica ese fenómeno. Dado que una base púrica se aparea siempre con la misma base pirimidínica (A – T y C – G), la cantidad de bases púricas será siempre igual a la de pirimidínicas, es decir, A + G = T + C ó A / T = C / G.
Las Copias para la Herencia: Duplicación del ADN
¿Cómo se copia el ADN? La clave está en la complementariedad de las bases nitrogenadas de las dos cadenas que lo forman. En 1958, M. Meselson (1930-) y F.W. Stahl (1929-) demostraron que a partir de una molécula de ADN se obtienen dos, cada una de las cuales porta una hebra del ADN original, en tanto que la otra hebra o cadena complementaria se va sintetizando siguiendo la complementariedad original (semiconservativa).
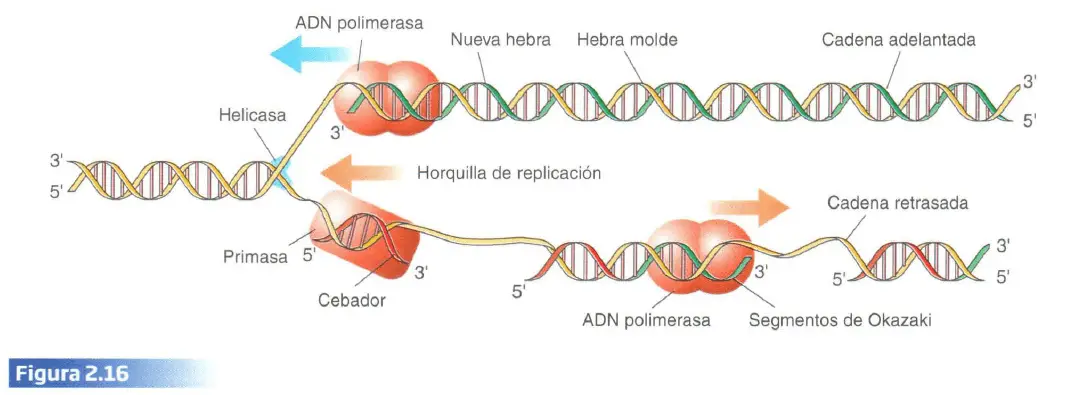
La replicación del ADN es llevada a cabo por un complejo enzimático en el que actúan numerosas enzimas, cada una de las cuales tiene una tarea concreta. Comienza en un punto en el que, con la participación de la enzima helicasa, las bases nitrogenadas son separadas y el ADN se desenrolla formándose la horquilla de replicación. La elongación de las nuevas hebras complementarias es catalizada por una ADN polimerasa. Esta enzima sólo puede leer la hebra molde en la dirección 3′ –>5′ por lo que la nueva cadena (en verde) sólo puede crecer en la dirección 5′–>3′ mediante la incorporación de los nuevos nucleótidos a su extremo 3′. Esta circunstancia tiene como consecuencia que, al avanzar el complejo enzimático en una dirección, sólo una de las cadenas puede ser leída en la dirección adecuada para la ADN polimerasa, 3′->5′ (la situada en la parte superior del gráfico), mientras que la otra, por ser antiparalela, tiene una orientación 5′->3′ en la dirección de avance del complejo enzimático y no puede ser leída directamente. Esto hace que la duplicación de esta hebra vaya retrasada con respecto a la otra (cadena adelantada) ya que su construcción se debe ir haciendo a cortos tramos (denominados segmentos de Okazaki) a medida que avanza el complejo enzimático. Estos segmentos se construyen gracias a que conforme se va abriendo la horquilla de replicación, la enzima primasa inserta un cebador sobre el que actúa la ADN polimerasa. Los tramos más antiguos son los que quedan más alejados de la punta de la horquilla de replicación y sirven de «tope» a los más recientes que al contactar con ellos son unidos por otra enzima del complejo de duplicación. Por supuesto, los nucleótidos que se incorporan a la nueva cadena son complementarios de la cadena original, según el principio C-G, T-A, de forma que la nueva hebra resultante es idéntica a la hebra complementaria original. Por tanto, las dos moléculas de ADN resultantes de la replicación son exactamente iguales que la molécula original: tienen la misma secuencia de bases nitrogenadas.
La afinidad sumamente específica entre entra las bases complementarias, que hace que sea enormemente difícil que sea una base inapropiada la que se empareje como complementaria en el proceso de replicación, hace del ADN, en efecto, una molécula química idónea para guardar la información genética.
LA INFORMACIÓN GENÉTICA
¿Cómo se codifica la información en el ADN? Los genes que codifican proteínas se denominan genes estructurales para diferenciarlos de aquellas otras secuencias de ADN que portan otro tipo de información.
En 1909, A.E. Garrad, publicó su trabajo «Errores congénitos del metabolismo», en el que señala que algunas enfermedades hereditarias son causadas por el efecto que la herencia ejerce sobre el metabolismo de determinadas sustancias. Garrad propone un nexo de unión ente genes y fenotipo: el metabolismo.
En 1941 , G. Beadle y E. Tatun, plantearon la hipótesis de un gen/un enzima. Esta hipótesis afirma que los genes regulan las características fenotípicas de los organismos gracias a que codifican la estructura de las enzimas que intervienen en todos y cada uno de los procesos metabólicos que acontecen en el organismo.
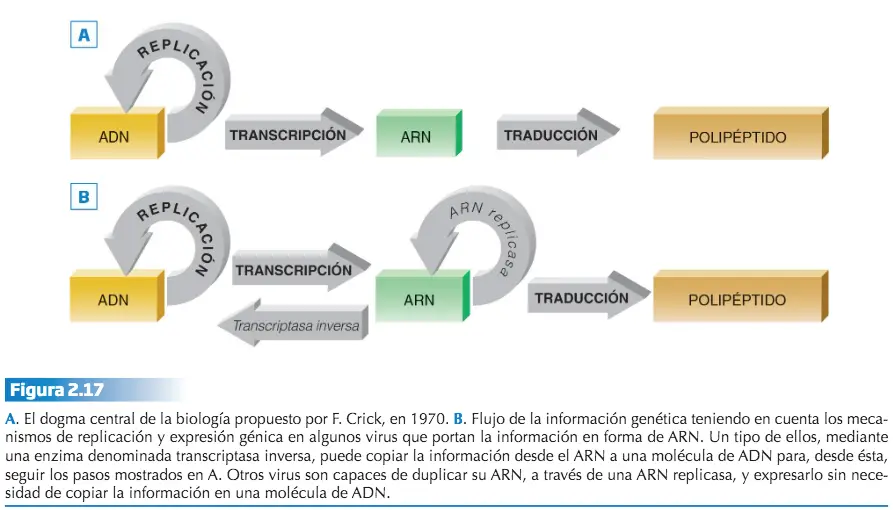
En 1970, Francis Crick, propuso el denominado dogma central de la Biología, que establece el flujo que sigue la información genética, la cual se halla en el ADN (molécula desde la que la información puede ser duplicada para su transmisión a otra célula, a través del proceso de replicación), de donde se trasfiere bioquímicamente a una molécula de ARN, mediante el denominado proceso de transcripción, y desde el ARN, a través del proceso de traducción, la información se expresa en una secuencia polipeptídica. Este dogma central ha tenido que ser ampliado en el sentido de que la información puede almacenarse en forma de ARN y trascribirse inversamente a ADN, siempre siguiendo el sistema de complementariedad de bases: es el caso de los retrovirus, que son virus cuya información genética se almacena en forma de ARN.
La Expresión Génica: la Información en Acción
La expresión génica es la manera en que la información codificada en el ADN se manifiesta en los procesos biológicos que dan lugar al desarrollo y funcionamiento característico de los seres vivos. Para ser efectiva, ha de seguir un proceso que consta de dos pasos, la trascripción y la traducción.
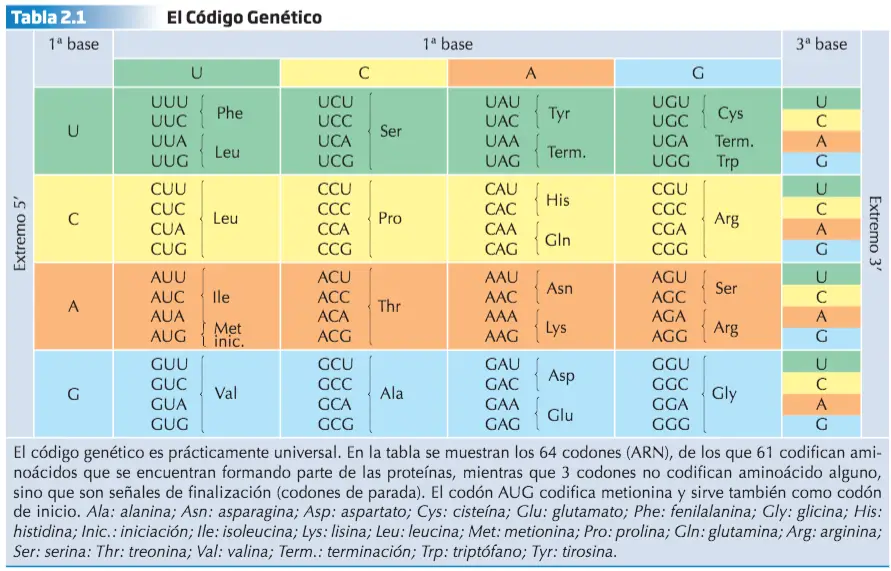
La Transcripción
El ADN de los eucariotas se encuentra situado en el núcleo celular, mientras que la maquinaria necesaria
para la síntesis de proteínas se halla en el citoplasma. El tamaño de la molécula de ADN y la importancia
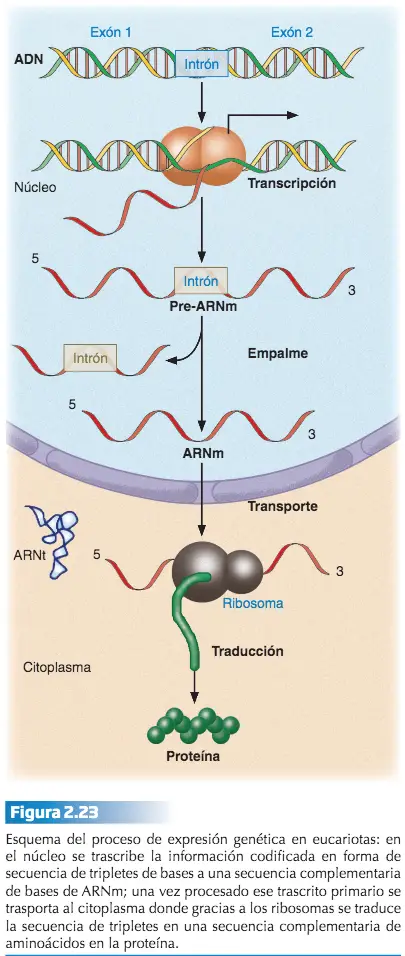
de la información en ella contenida pueden ser dos de los motivos que hacen que el ADN no viaje hasta el citoplasma para transmitir las instrucciones necesarias para la síntesis proteica. Por ello, cada vez que es necesaria la producción de un determinado polipéptido, la información de su secuencia de aminoácidos es copiada desde el correspondiente gen a un ácido ribonucleico. A este proceso se le denomina transcripción.
El ARN formado es el que viaja hasta el citoplasma transportando la información para que el polipéptido en cuestión sea sintetizado. Por este motivo a ese ARN se le llama ARN mensajero (ARNm). El proceso de transcripción es catalizado por una enzima perteneciente al grupo de las ARN polimerasas.
En la transcripción se siguen las reglas de complementariedad, con la salvedad de que en vez de añadir un nucleótido de timina cuando en la hebra molde de ADN aparece un nucleótido de adenina, se añade un nucleótido de uracilo en la cadena de ARN en crecimiento. La ARN polimerasa se une a una región específica situada por delante del gen que se va a transcribir, llamada promotor, y desde esta región inicia la síntesis del ARNm. La transcripción del ARN finaliza cuando la ARN polimerasa alcanza una región específica del ADN situada al final del gen, denominada secuencia de fin. No todas las secuencias de ADN guardan información referente a la estructura primaria de los polipéptidos. Otros segmentos de ADN se transcriben a ácidos ribonucleicos con funciones distintas a la del ARNm. Son los ácidos ribonucleicos ribosómicos (ARNr), que forman parte del ribosoma, y los ácidos ribonucleicos de transferencia (ARNt), que se encargan de transportar los aminoácidos durante la síntesis de proteínas.
Maduración del ARN
En algunos procariotas y en prácticamente todos los eucariotas, los ARNm experimentan una modificación de su estructura una vez sintetizados.
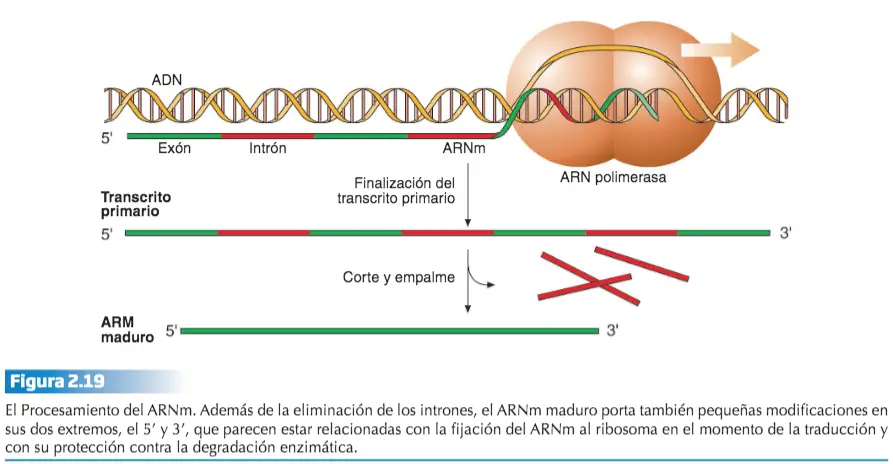
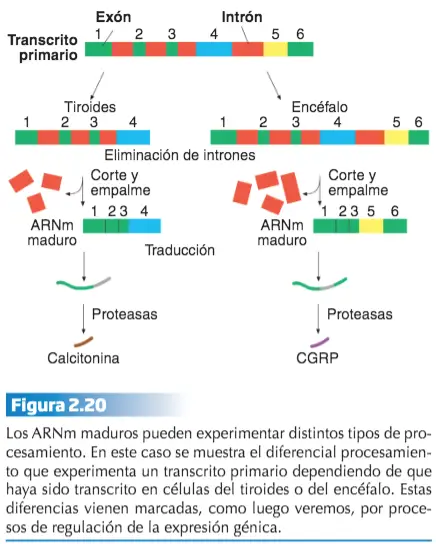
El ARNm que produce la ARN polimerasa se denomina transcrito primario. El transito primario porta la secuencia que codifica el polipéptido, sin embargo, esta secuencia no está colocada de forma continua en este ARNm.
- Intrones. Secuencias intercaladas a lo largo del transcrito primario, separadas por segmentos no codificantes.
- Exones. Secuencias intercaladas a lo largo del transcrito primario que contienen la información para producir la proteína codificada en el gen, separadas por segmentos no codificantes.
En los eucariotas, los intrones representan un% mayor de la secuencia génica que el dedicado a los exones. A través de un proceso de corte y empalme (splicing) denominado maduración o procesamiento del transcrito primario, se eliminan los intrones y se colocan secuencialmente los exones, obteniéndose un ARNm maduro que porta la secuencia lineal de un polipéptido funcional.
Dependiendo de los genes, hay transcritos primarios que tras su procesamiento codifican siempre el mismo polipéptido, y otros que pueden experimentar varios tipos de maduración que originan polipéptidos distintos, en función de la célula en que se exprese, y la etapa de desarrollo en que se encuentre el organismo.
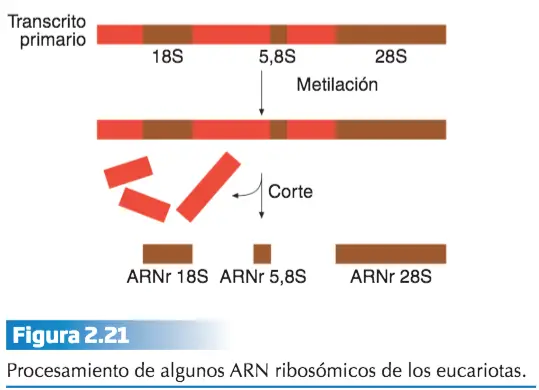
Los ARN ribosómicos y de transferencia también experimentan maduración. Así, en eucariotas, los ARNr 18S, 28S y 5,8S proceden de un solo transcrito primario que tras su maduración origina esos distintos ARNs ribosómicos .
El Lenguaje de la Vida: El Código Genético
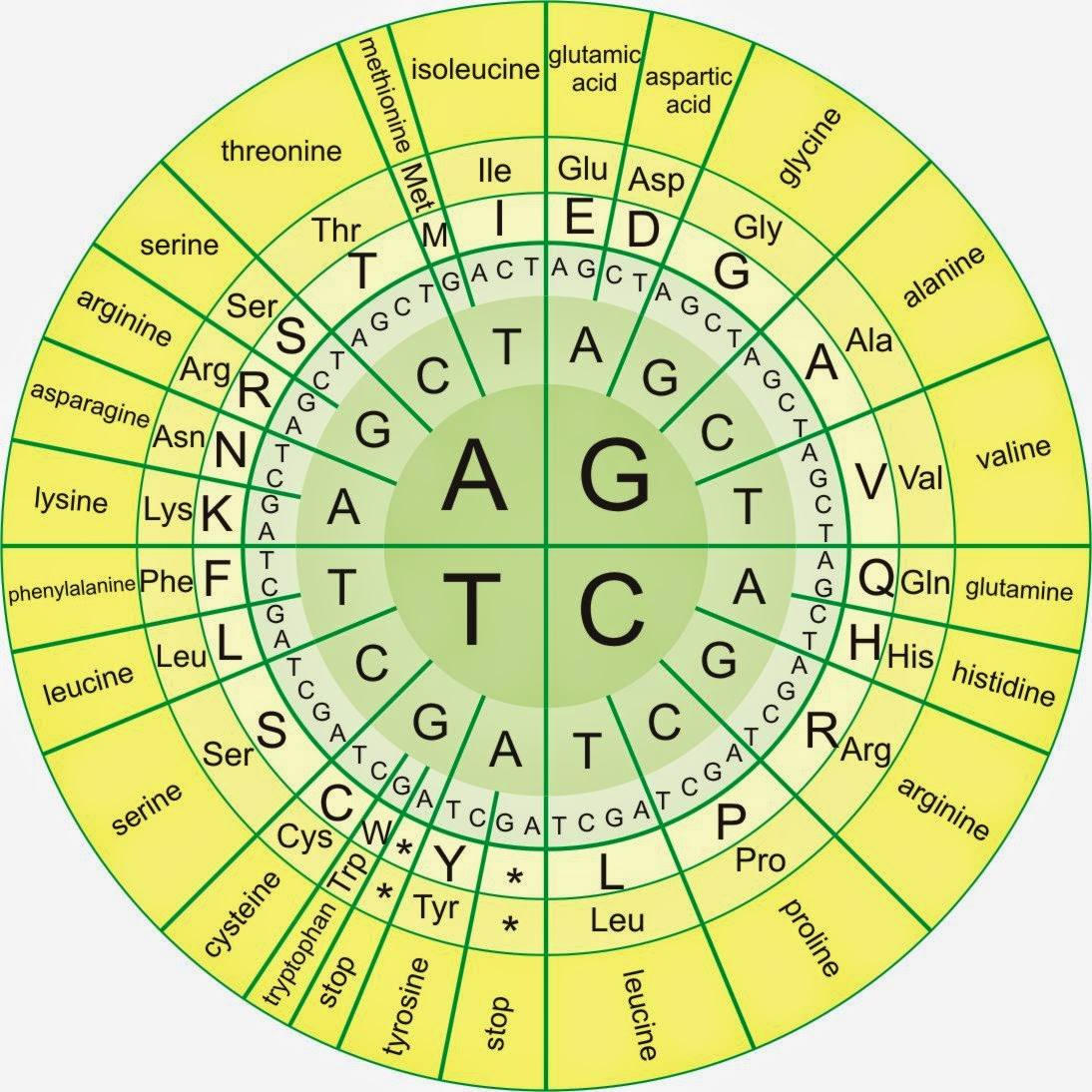
¿Cómo está codificada en el ADN la información referente a la secuencia de aminoácidos de un polipéptido? Es sabido que todas las proteínas están constituidas por cadenas de aminoácidos, de los que sólo se usan 20, así que los polipéptidos (proteínas) se diferencian unos de otros sólo por el orden en que estén unidos los aminoácidos que los constituyen. El ADN, por su parte, contiene la información acerca de las secuencias de aminoácidos de todos los polipéptidos del organismo. Dado que la naturaleza del ADN y la de los polipéptidos son distintas, esa información debe ser guardada de forma cifrada de acuerdo con un código. El ADN está formado por sólo 4 nucleótidos cuya diferencia estriba en las bases que los constituyen, Adenina, Timina, Guanina y Citosina, es decir, la información que porta utiliza un alfabeto de cuatro letras (A, T, G y C).
Sin embargo, los polipéptidos utilizan 20, y cabe preguntarse cómo es posible que con las 4 letras del ADN se pueda codificar la información relativa a 20 aminoácidos diferentes para formar los polipéptidos. Para encontrar la solución a esta pregunta, y asumiendo que el orden de las bases en el ADN determina el orden de los aminoácidos en las proteínas, los científicos emplearon como primera aproximación la lógica de las técnicas criptográficas para proponer una hipótesis.
Si tomamos de una en una las cuatro bases el ADN sólo podría guardar información acerca de cuatro aminoácidos. Si el código se estableciese combinando esas cuatro bases de dos en dos, se podrían formar 42 combinaciones posibles, es decir, el ADN podría guardar información acerca de 16 aminoácidos. Si combinamos esas «letras» de tres en tres, se podrán formar 64 combinaciones. Con 20 hubiese bastado, pero como veremos, las combinaciones sobrantes también tienen un significado.
Durante los primeros años de la década de 1960 y gracias a los datos experimentales aportados por los
grupos de trabajo dirigidos por M. Nirenberg, Severo Ochoa y H.G. Khorana, se comprobó que la base del
código genético es el triplete (en el ADN), o codón (cuando nos referimos a ese triplete en el ARNm). Está constituido por una secuencia cualquiera de los tres nucleótidos de los cuatro posibles (de adenina, guanina, citosina y timina; el uracilo sustituye a la timina en el ARNm). El orden en que van los tripletes especifica el orden en que van los aminoácidos en las proteínas.
Para entender bien cómo funciona el código genético, hay que entender las siguientes propiedades:
- A) Es redundante o degenerado: Cada aminoácido puede estar codificado por más de un codón. Habiendo 64 tripletes posibles y sólo 20 aminoácidos, sobran tripletes, cada aminoácido puede estar codificado por más de un triplete, es decir, hay tripletes «sinónimos».
- B) Es un código sin superposición: Un nucleótido sólo pertenece a un codón y no a varios. Por ejemplo, en la secuencia AUGCAUAAG, los codones serían: AUG, CAU, AAG y no UGC, AUA, GCA o UAA.
- C) La lectura es lineal y continua: La lectura del ARNm se inicia en un punto y avanza de codón en codón sin interrupciones ni saltos.
- D) Es universal: Prácticamente todos los seres vivos, desde una bacteria a un mamífero, pasando por las plantas o los hongos, utilizan el mismo código para traducir el mensaje del ADN a polipéptidos.
La Traducción
El proceso mediante el cual la información contenida en el ARNm, en un alfabeto de cuatro letras, es convertida, siguiendo las reglas del código genético, al alfabeto de 20 letras de los polipéptidos se le denomina traducción.
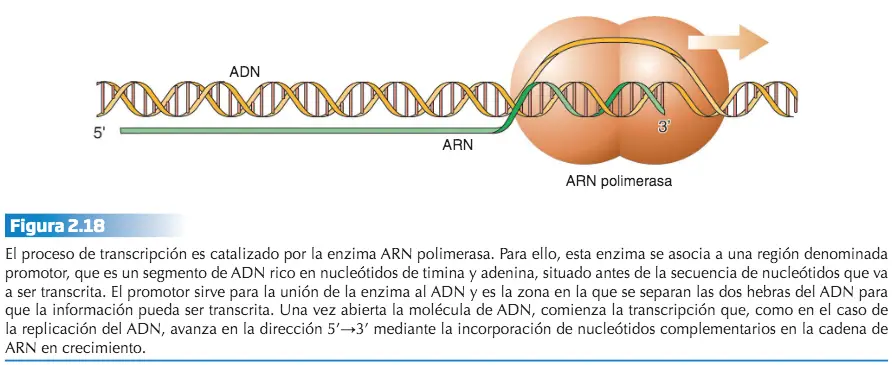
La síntesis del polipéptido cuya secuencia lleva cifrada el ARNm se inicia en los ribosomas. A través de un proceso enzimático, los ácidos ribonucleicos de transferencia (ARNt) van incorporando los correspondientes aminoácidos especificados por la secuencia lineal de codones del ARNm. Esto se consigue gracias a que existen tantos ARNts como codones distintos puede haber en el ARNm. La diferencia entre los ácidos ribonucleicos de transferencia radica en el triplete de nucleótidos complementario de cada uno de los codones del ARNm, denominado anticodón, y en el aminoácido que transporta, que no es otro que el especificado por su codón complementario. El resultado es la formación de un polipéptido con una función biológica concreta y distinta de la de cualquier otro cuya secuencia de aminoácidos sea diferente.
REGULACIÓN DE LA EXPRESIÓN GÉNICA
La mitosis asegura un reparto completo y equitativo de la información genética. Por este motivo, todas las células de un individuo portan la misma información, tienen idénticos genes en sus núcleos. Dentro de la célula ya diferenciada, el metabolismo celular varía continuamente a lo largo de su ciclo vital. Distintas rutas de síntesis (anabolismo) o de degrada ni simultánea para todos los genes, sino que se activa sólo cuando los correspondientes polipéptidos se necesitan.
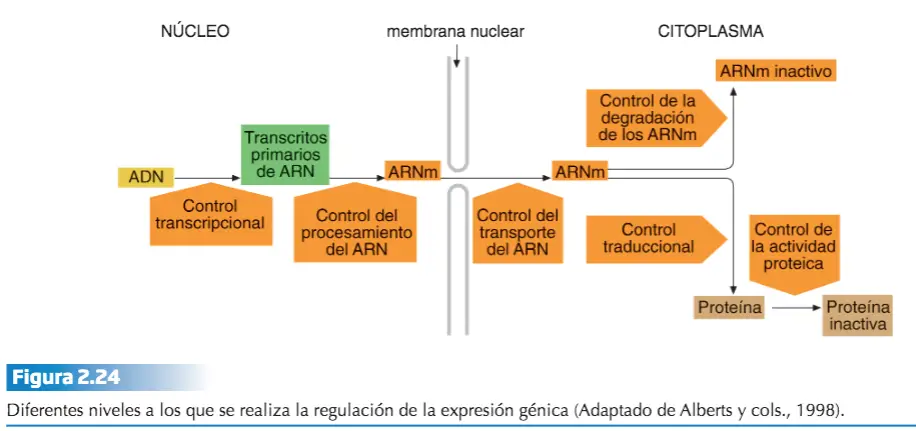
Todo ello pone de manifiesto que la expresión génica está regulada de forma precisa, tanto durante las sucesivas etapas del desarrollo del organismo, como a lo largo del ciclo vital celular. Podemos distinguir una regulación a corto plazo y otra a largo plazo.
- Regulación a corto plazo. Está relacionada con el metabolismo celular, y provoca cambios en el ADN que alteran de forma pasajera la expresión génica.
- Regulación a largo plazo. Está relacionada con el desarrollo del organismo y conduce a cambios en el ADN de la célula que conllevan el bloqueo permanente (aunque no irreversible) de la expresión de determinados genes.
Los núcleos celulares son totipotentes, es decir, la información contenida en ellos es similar y es susceptible de volver a expresarse de nuevo. En esta circunstancia se encuentra la base de los experimentos de clonación. Puesto que en cualquier célula de un organismo está toda la información genética del mismo, si extraemos el núcleo de una célula y lo introducimos en un ovocito al que se le ha eliminado el suyo, es decir, su información genética, el desarrollo de esa nueva célula originará un individuo genéticamente
idéntico al donador del núcleo trasplantado, esto es, un clon.
Regulación de la Expresión Génica a Corto Plazo
- Está relacionada con el control del metabolismo celular.
- Produce alteraciones pasajeras de la expresión génica.
- Genes reguladores, codifican la secuencia de las denominadas proteínas reguladoras o factores de transcripción. Estas proteínas reguladoras pueden, o bien impedir, o bien inducir (activar), la expresión de los genes estructurales.
- Se unen al inicio de los genes estructurales, en la secuencia reguladora, impidiendo o facilitando la unión de la ARN polimerasa y, por tanto, la expresión del gen estructural.
- En algunos casos, la conformación espacial adecuada para que la proteína pueda unirse a la secuencia reguladora depende de la interacción que establezca con otras moléculas denominadas correpresores e inductores.
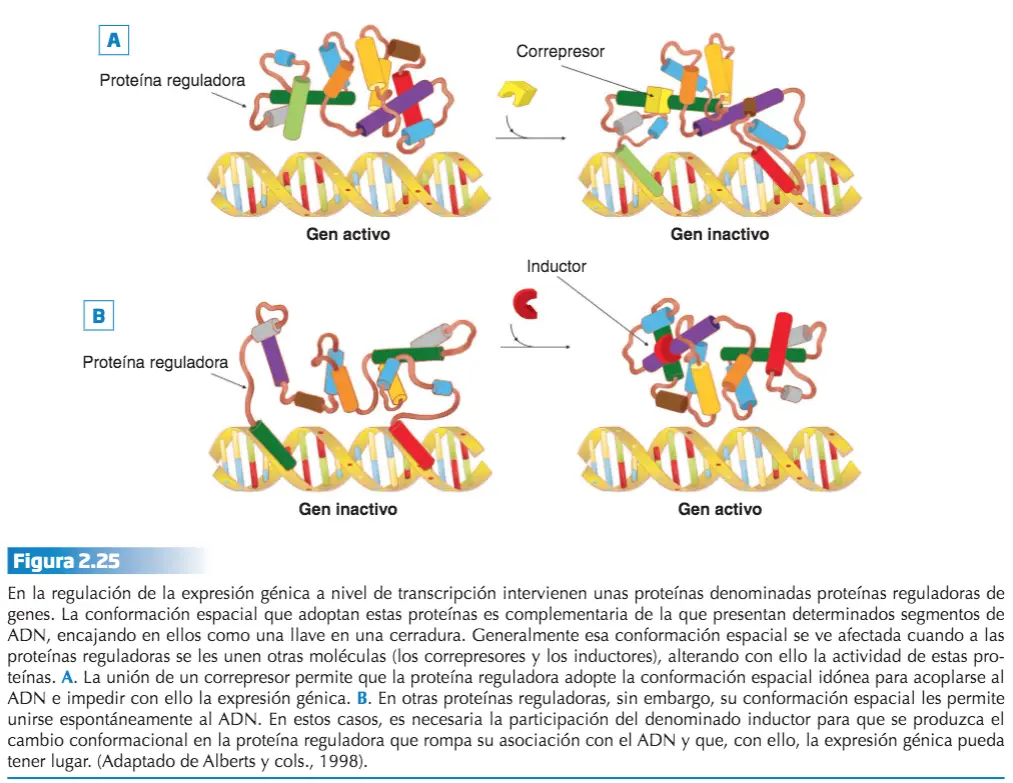
- Los correpresores son moléculas a las que necesitan acoplarse algunas proteínas reguladoras para adoptar la conformación espacial adecuada que les permita unirse a una secuencia reguladora concreta del ADN, e impedir (reprimir) la expresión de un gen.
- Un caso especial de correpresores es el denominado ARN de interferencia (ARNi ). El ARNi bloquea la expresión de genes con una extraordinaria especificidad y se ha visto que desempeña una función esencial en la regulación del desarrollo y plasticidad neuronales.
- El efecto represor del ARNi se ejerce principalmente por la acción conjunta de un ARN de doble hebra o ARNdh y la formación de un complejo multiproteico que tiene como resultado final la inhibición del proceso de traducción del ARNm al que se haya acoplado el ARNdh.
- El ARN de doble hebra es transcrito del ADN a partir de unos pequeños genes denominados microARN (miARN),
- Los genes microARN (miARN) no codifican proteínas.
- Se ha conseguido generar ARNdh sintéticos que se emplean para explorar posibles nuevas mejoras terapéuticas.
- Los inductores son moléculas que, al unirse a las proteínas reguladoras, hacen que éstas no puedan unirse al ADN, permitiendo (induciendo) que el gen pueda ser transcrito.
- La separación del inductor de la proteína reguladora hace que la unión con el ADN se pueda establecer y con ello la represión de la expresión génica.
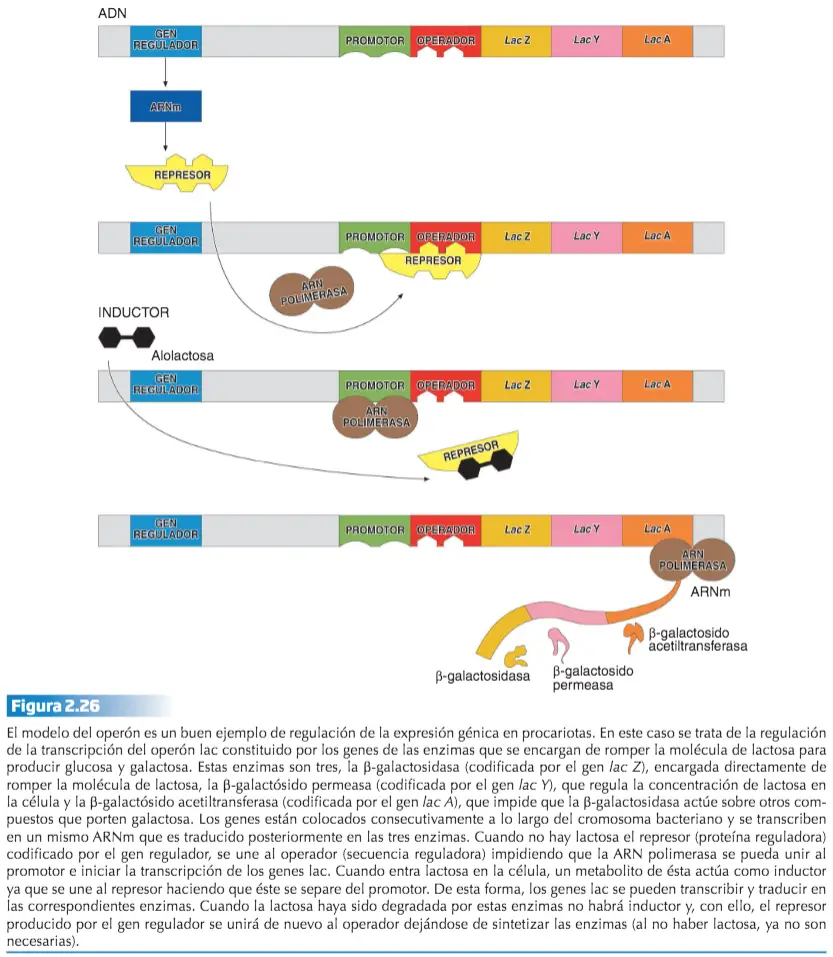
El modelo del operón de Jacob y Monod (1961). Explica la síntesis de la enzima encargada de degradar la lactosa ? galactosidasa.
- Cuando no hay lactosa el represor (proteína reguladora) codificado por el gen regulador, se une al operador (secuencia reguladora) impidiendo que la ARN polimerasa se pueda unir al promotor e iniciar la transcripción de los genes lac.
- Cuando entra lactosa en la célula, un metabolito de ésta actúa como inductor ya que se une al represor haciendo que éste se separe del promotor, permitiendo que actúe la ARN polimerasa.
Regulación de la Expresión Génica a Largo Plazo
La diferenciación celular junto con la compleja organización pluricelular que da lugar a los distintos órganos del cuerpo y hace que éste adopte su forma tridimensional típica necesita de procesos regulatorios relativamente prolongados: regulación génica a largo plazo. Los mecanismos implicados no se conocen bien, hay bastantes datos que apuntan a complejas interacciones entre diferentes grupos de genes y distintos tipos de moléculas durante el desarrollo embrionario. Entre estos genes se encuentran los denominados homeogenes, o genes homeobox, así llamados por tener en común una secuencia característica de 180 bases (que codifican una secuencia de 60 aminoácidos) en uno de sus extremos. Las proteínas codificadas por estos genes regulan la ex presión de genes que poseen elementos que responden a esta secuencia homeobox.
En la diferenciación celular están involucrados también otros mecanismos de inactivación génica permanente, como la metilación y la condensación del ADN.
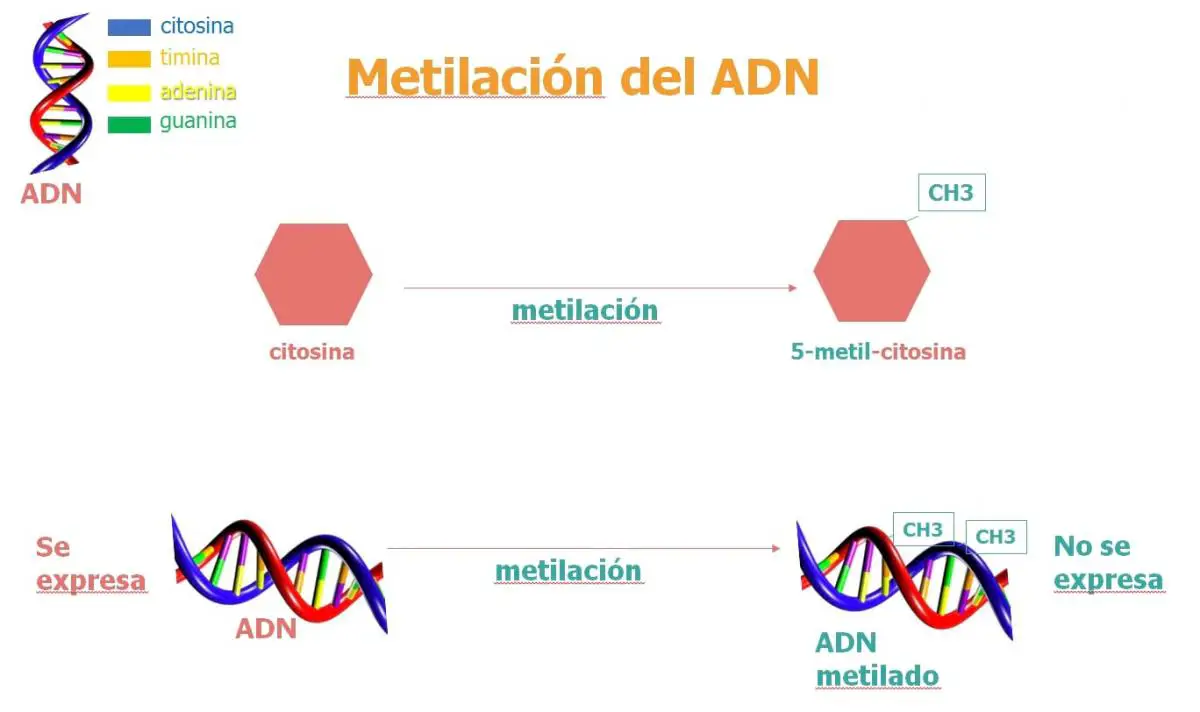
- Metilación: La metilación del ADN es una reacción catalizada enzimáticamente mediante la cual se inserta un grupo metilo (-CH3) en la base nitrogenada de los nucleótidos (sobre todo afecta a los de citosina) que provoca un cambio que hace la enzima ARN polimerasa no pueda unirse.
- Condensación: la condensación del ADN impide que la ARN polimerasa pueda acceder a los respectivos promotores, existiendo una relación inversa entre el grado de condensación del ADN y el proceso de transcripción. La condensación afecta a grandes segmentos de ADN o a cromosomas enteros.
Tanto la metilación como la condensación, parecen estar implicadas en los procesos de diferenciación celular. Estos procesos suelen suceder en las primeras etapas del desarrollo y una vez que se han producido, tanto las zonas metiladas, como las altamente condensadas, se heredan a través de la mitosis.
Aunque los factores que hemos descrito en relación la regulación a corto plazo de la expresión génica en su día fueron conceptualizados como factores epigenéticos, hoy en día el término epigenética hace referencia especialmente a factores heredables, bien que de tipo más o menos transitorio, en los que no se dan cambios en el ADN: en realidad son cambios que pueden pasarse a la siguiente generación y que tienen que ver con mecanismos a largo plazo de regulación de la expresión génica.
LOS ERRORES QUE NOS MATAN Y NOS HACEN EVOLUCIONAR: LA MUTACIÓN
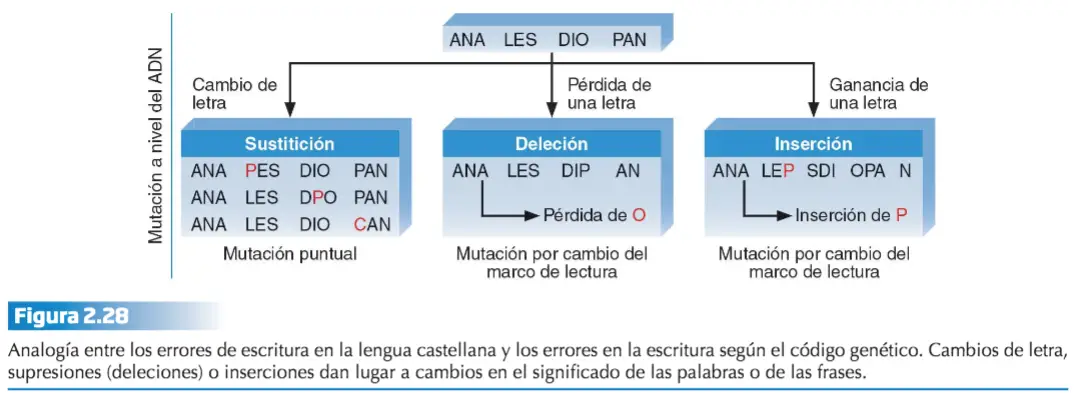
El término «mutación» es introducido en 1901 , por Hugo De Vries (1848-1935), y hace referencia a cualquier cambio permanente en el material génico no debido a la segregación independiente de los cromosomas o a la recombinación que ocurre durante el proceso de meiosis. Una mutación es cualquier alteración en la secuencia de nucleótidos del ADN, alteración que puede suponer simplemente, la inserción o deleción de un par de bases, o bien la sustitución de un par de bases por otro (mutaciones puntuales), pero también la inserción, deleción, sustitución o cambio de orientación de segmentos más o menos grandes de ADN del cromosoma.
Las mutaciones se clasifican según diferentes criterios:
- Por sus causas:
- Espontáneas. Por fallos en la copia del ADN.
- Inducidas por agentes mutágenos, sustancias cancerígenas, radiaciones (rayos X, rayos gamma, ultravioleta), etc.
- Por el tipo de células donde ocurre:
- Somáticas.
- Germinales. Estas últimas son las que pueden pasar a la siguiente generación, es decir, las verdaderamente heredables, puesto que se ubican en el ADN de los gametos.
- Por el cromosoma donde tienen lugar:
- Autosómicas. Ligadas al cromosoma X (o al Y).
- Genómica, el cambio en el número de cromosomas:
- aneuploidías por pérdida (monosomía) o ganancia (trisomía) de un cromosoma.
- poliploidías, cuando hay más de dos copias de cada cromosoma en el genoma de un individuo.
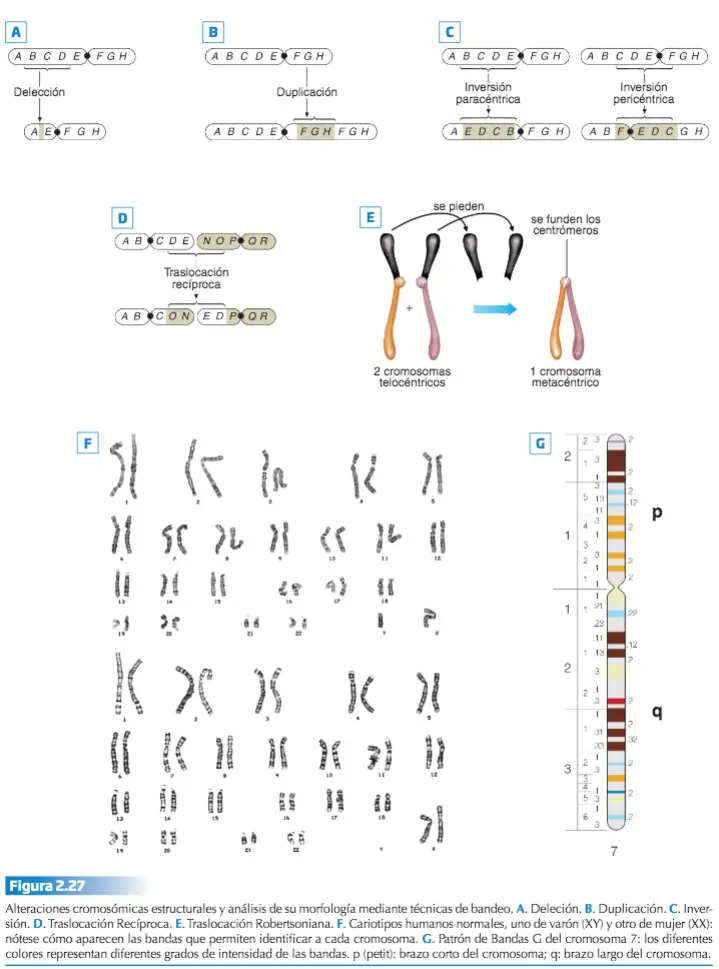
- Según el cambio molecular que produce en el ADN.
- Puntuales o de sustitución de un único nucleótido, lo que da lugar a polimorfismo de un solo nucleótido (SNP). A veces se considera SNP la inserción o deleción de un nucleótido, pero en este caso, en realidad, ha de considerarse una mutación de marco o patrón de lectura (frameshift). Para que se considere polimorfismo, la variante ha de estar presente en al menos el 1 % de la población; por eso a veces se prefiere hablar de variante de un solo nucleótido.
- De marco (frameshit), al insertarse o suprimirse uno o varios, pares de nucleótidos en la secuencia se altera el patrón de lectura de tripletes.
Pueden producir cambios en el fenotipo, a nivel bioquímico, fisiológico o conductual. También puede ocurrir, que un trozo completo de ADN se duplique, de forma que en un mismo cromosoma haya dos copias de un gen.
- Basada en sus efectos fenotípicos (considerada como la más importante).
- Modificación de los tripletes, o de la secuencia de tripletes de los genes:
- Con pérdida de función, bien porque la secuencia resultante tras la mutación no sea legible (no codifique nada), o porque la nueva proteína tenga poca o ninguna funcionalidad. Estas mutaciones de pérdida de función pueden ser recesivas o dominantes: serán dominantes cuando en heterocigosis el alelo normal no sea suficiente para permitir un adecuado funcionamiento, o bien, cuando el alelo mutado codifique una proteína que impide el funcionamiento apropiado del producto del alelo no mutado.
- Con ganancia de función, si el producto del gen mutado es más activo en sus funciones o adquiere otras nuevas: esto podría ocurrir si el nuevo alelo codifica una proteína con diferente secuencia de aminoácidos. También podría ocurrir que la mutación afecte a las regiones reguladoras del gen, haciendo que se exprese en mayor medida de lo que lo hace el gen no mutado. Estas mutaciones con ganancia de función suelen ser dominantes.
- Mutación letal. Cuando una mutación da lugar a la interrupción de un proceso biológico esencial para la vida (en realidad basta con que la mutación provoque esterilidad en el sujeto portador para que, en sentido evolutivo, dicha mutación sea letal).
- Modificación de los tripletes, o de la secuencia de tripletes de los genes:
La mayoría de las mutaciones son neutras; igualmente serán neutras si no alteran la estructura de la proteína o los niveles de expresión del gen: cuando el triplete alterado por una mutación puntual da lugar en la proteína al mismo aminoácido original se habla de sustitución silenciosa o sinónima.
Las mutaciones generan la variabilidad necesaria para que la selección natural pueda actuar. Ocurren al azar, y en humanos las mutaciones puntuales ocurren con una tasa de 1,20 X 10-8 por base y generación (un cambio por cada 100 millones de bases). Sin embargo, el número de polimorfismos de un solo nucleótido encontrado en los genomas humanos secuenciados es de 3-4 millones; de hecho esto representa uno por cada 1000-1200 bases. Los SNP son mucho más frecuentes en los intrones (segmentos de ADN que no se traducen a proteínas) que en los exones.
COMPLEMENTOS DE GENÉTICA MENDELIANA
Los experimentos de Mendel demostraron que la herencia genética se trasmite en unidades concretas (genes), de forma que tales unidades no se diluyen o mezclan para dar lugar a un fenotipo intermedio en los descendientes
Variación de la Dominancia e Interacciones Génicas
Herencia intermedia
Si Mendel hubiera utilizado en su investigación la planta Mirabilis jalapa (dondiego de noche) hubiera encontrado fenotipos intermedios:
Cuando se cruzan dos razas puras de esta planta, una de flores rojas y otra de flores blancas, en la generación F1 todas las flores son rosas.
La autofecundación de la generación F1 da lugar a una generación F2 donde las proporciones son 1:2:1 (blancas; rosas; rojas), donde las blancas y rojas son homocigóticas para uno de los dos alelos, mientras que las rosas son heterocigóticas.
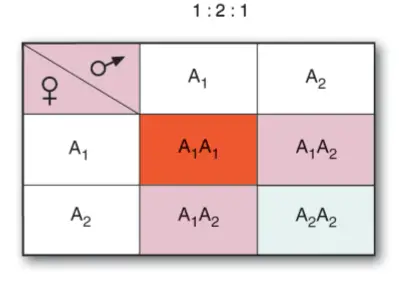
La explicación en este caso, estriba en el hecho de que uno de los alelos, A1 , aporta pigmento, mientras que el alelo A2 no aporta pigmento. Es
importante notar que el fenotipo depende de cuántos alelos de un tipo u otro haya en el genotipo: si hay dos alelos A1 (genotipo A1A1) , la cantidad de pigmento rojo es el doble que si el genotipo es heterocigótico.
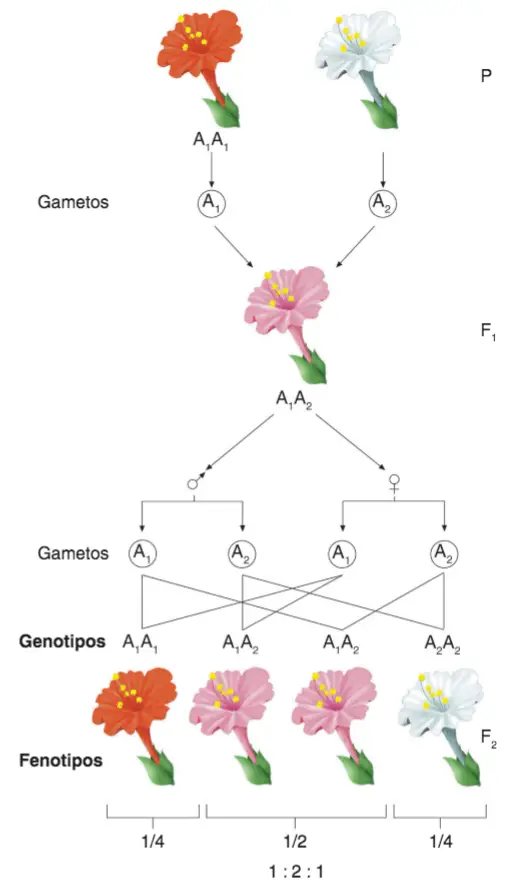
La F2 produce una proporción 1:2:1 en vez de la 3:1, habitual en los casos de dominancia.
Las flores del experimento de Correns (Mirabilis jalapa)
El botánico Correns realizó un experimento con flores de la planta comúnmente llamada Dondiego de noche, la cual posee variedades de flores completamente rojas o completamente blancas. Correns realizó cruces entre plantas homocigotas de color rojo y plantas homocigotas de color blanco; la descendencia presentó un fenotipo intermedio al de los parentales (color rosado). El alelo de tipo salvaje para el color de la flor roja se designa (A1A1) y el alelo blanco es (A2A2). Así:
- Generación parental (P): (A1A1) (flores rojas) x (A2A2) (flores blancas).
- Generación filial 1 (F1): A1A2 (flores rosadas). Al permitir que estos descendientes F1 se auto-fertilizaran, la siguiente generación (F2) producía 1/4 de plantas con flores rojas, 1/2 de plantas con flores rosas y 1/4 de plantas con flores blancas. Las plantas rosas en la generación F2 eran heterocigotas con fenotipo intermedio.
- La generación F2 mostró una relación fenotípica 1: 2: 1, que era diferente de la relación fenotípica 3: 1 observada para la herencia mendeliana simple.
Lo que ocurre en el ámbito molecular es que el alelo que causa un fenotipo blanco da como resultado la falta de una proteína funcional, requerida para la pigmentación. Dependiendo de los efectos de la regulación genética, los heterocigotos pueden producir solo el 50 % de la proteína normal. Esta cantidad no es suficiente para producir el mismo fenotipo que el homocigoto A1A1, que puede producir el doble de esta proteína. En este ejemplo, una explicación razonable es que el 50 % de la proteína funcional no puede lograr el mismo nivel de síntesis de pigmento que el 100 % de la proteína.
Análisis del Fenotipo a Diferentes Niveles: Codominancía, Dominancia Incompleta y Pleitropismo La conexión entre genotipo y fenotipo se explica
La interpretación de dominancia y recesividad no son suficientes para describir totalmente la conexión entre genotipo y fenotipo. En los grupos sanguíneos A, B y 0 donde los sujetos heterocigóticos para los alelos A y B (genotipo AB) presentan un fenotipo doble respecto a los homocigóticos AA y BB, tanto el alelo A como el alelo B son funcionales y su expresión da lugar a que en los glóbulos rojos de los sujetos AB se formen tanto el antígeno A como el antígeno B. Los sujetos AA, A0, BB y B0 sólo producen una de las dos variantes posibles del antígeno (el alelo 0 es recesivo porque su presencia no genera ningún tipo de antígeno).
Codominancia. La codominancia es una relación entre dos versiones de un mismo gen. Los individuos reciben una versión de un gen, llamada alelo, de cada progenitor. Si los alelos son diferentes, normalmente se expresará el alelo dominante, mientras que el efecto del otro alelo, llamado recesivo, queda enmascarado. Pero cuando hay codominancia, entonces ningún alelo es recesivo y el fenotipo de ambos alelos es expresado. La codominancia significa que ningún alelo puede enmascarar la expresión del otro alelo. Un ejemplo lo tenemos en humanos sería el grupo sanguíneo AB0, en el que ambos alelos A y B se expresan. Por tanto, si un individuo hereda el alelo A de su madre y el alelo B de su padre, tendrá el tipo sanguíneo AB.
Dominancia incompleta. La dominancia incompleta es el fenómeno genético en el cual el alelo dominante no enmascara el efecto del alelo recesivo por completo; es decir, que no es completamente dominante. La dominancia incompleta fue descrita por primera vez en 1905 por el botánico alemán Carl Correns, en sus estudios del color de las flores de la especie Mirabilis jalapa. El efecto de la dominancia incompleta se hace evidente cuando se observan los descendientes heterocigotos de un cruce entre homocigotos. En este caso, los descendientes poseen un fenotipo intermedio al de los parentales y no el fenotipo dominante, que es lo que se observa en los casos donde la dominancia es completa. Un alelo muestra dominancia cuando suprime la expresión o domina los efectos del alelo recesivo.
Pleiotropismo. El pleiotropismo aparece cuando un solo gen afecta a más de un fenotipo, es decir, a más de una característica fenotípica (por ejemplo el color de los ojos, el color del pelo, la altura, las pecas, etc.). Un mismo gen puede estar involucrado en varios rasgos fenotípicos, es lo que se conoce como pleiotropismo, y además, cada alelo puede resultar dominante en alguno de esos rasgos y recesivo en otros.
Epistasia
La epistasia o epistasis se debe a que la expresión de uno de los dos genes es requisito imprescindible para que se manifieste el efecto sobre el fenotipo del otro gen. Por ejemplo:
El fenotipo Bombay: A nivel molecular el grupo sanguíneo A es debido a la acción de un enzima que añade el polisacárido A a un polímero de azúcar conocido como sustancia H, mientras que el grupo B es el resultado de una variante de la misma enzima que añade al polímero H el polisacárido B. El tipo 0 se debe a que no se sintetiza ninguna de las dos formas de enzima y por tanto, no se añade nada al polímero H. Sucedió en Bombay que una mujer del grupo 0 tuvo con un hombre del grupo A una hija del grupo AB y un hijo del grupo B, cosa que no es posible basándonos en el modo de herencia de un solo gen con dos alelos. Se demostró que la mujer aparentaba ser del grupo 0 porque en realidad no producía el polímero H (para este segundo gen, el genotipo de la mujer era homocigótico recesivo hh): sus hijos sí recibieron el alelo H del padre, lo que hizo que se manifestara el verdadero grupo sanguíneo de la madre, que de haberse podido manifestar hubiera sido B o AB.
TIPOS DE TRANSMISIÓN GÉNICA Y CONDUCTA HUMANA
Herencia monogénica. Los rasgos de un organismo pueden estar determinados por un único gen, como es el caso del color de la flor del guisante o del albinismo.
Herencia poligénica. Los rasgos de un organismo pueden estar determinados por varios genes como ocurre con la altura de una persona o su inteligencia.
Por cuestiones obvias no es posible experimentar con seres humanos, la alternativa es el estudio del patrón de transmisión del carácter. Dicho patrón se establece a través de la información recogida de la familia en la que se detecta el carácter de interés. Esta información se suele resumir representándola en forma de lo que se denomina una genealogía o pedigrí.
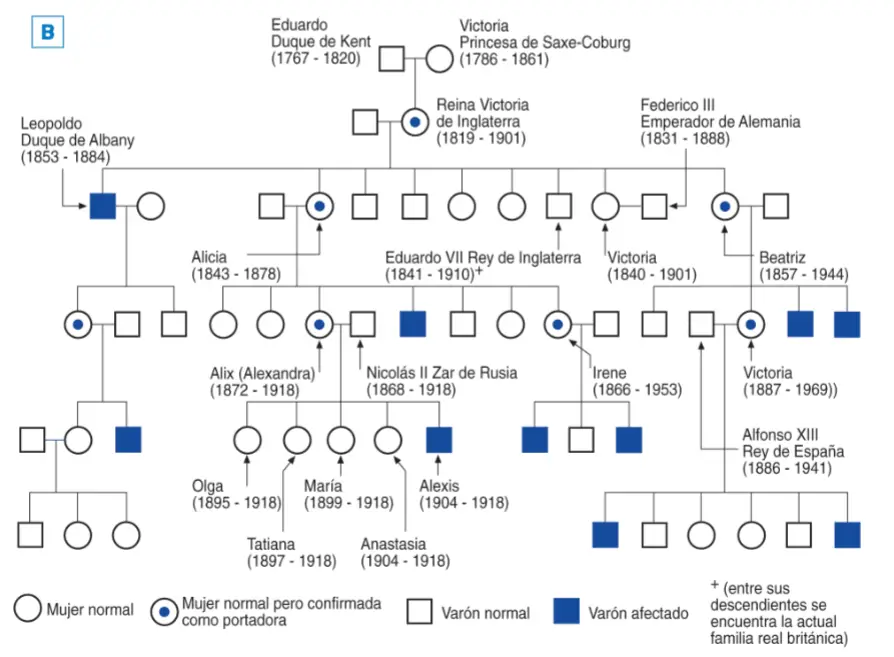
hemofilia en el árbol genealógico de la reina Victoria de Inglaterra.
Los patrones de transmisión de un carácter mendeliano, detectados a través de las genealogías, dependen
de dos factores:
- El tipo de cromosoma donde se halla el gen implicado. La localización puede ser:
- Autosómica, cuando el locus se halla en un autosoma (denominación que se da a cualquiera de los cromosomas que no sean los sexuales),
- ligada a los cromosomas sexuales.
- El tipo de expresión fenotípica de las variantes alélicas del gen, ésta puede ser diversa, pero en la mayoría de casos es dominante o recesiva
Por tanto, según estos criterios, se establecen tres tipos de patrones de transmisión en la herencia monogénica: autosómica dominante, autosómica recesiva y ligada al sexo.
Transmisión Autosómica Dominante
El gen FOXP2 y el lenguaje
Pedigrí de la familia KE. Los cuadrados representan a los varones y los círculos a las mujeres. La diagonal señala que esos sujetos han muerto. Los símbolos sombreados representan a sujetos con problemas en el habla y de tipo lingüístico. Los asteriscos señalan a los sujetos a los que no se pudo estudiar genéticamente.
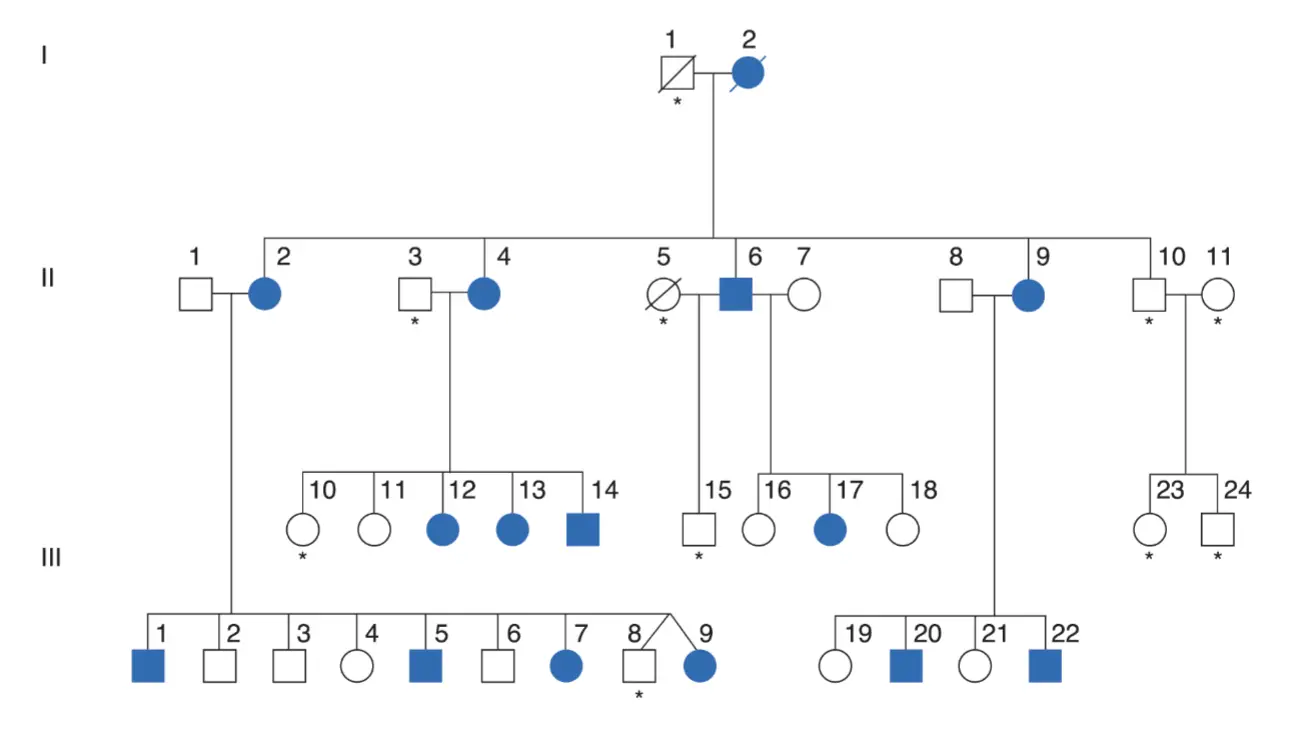
Muchos de los miembros de esta familia presentaban una notable alteración en sus capacidades lingüísticas unida a dispraxia verbal. Este fenotipo es compatible con la acción de un alelo dominante. Estudios moleculares complementarios demostraron que los individuos afectados, y los que no lo estaban, eran portadores de una mutación sin sentido del gen conocido como FOXP2. Otras mutaciones observadas de este gen, concretamente una mutación puntual sin sentido en otra familia diferente, también ocasionara un fenotipo lingüístico similar, no deja duda acerca de la relación entre genotipo y fenotipo en este locus del gen FOXP2.
El gen FOXP2 no es privativo de la especie humana, se halla también en el genoma de las aves y todos los mamíferos. Sólo hay tres sustituciones de aminoácidos de la proteína que codifica este gen, que diferencien entre ratones y humanos, y sin embargo sólo 1 entre ratones y primates no humanos; esto implica dos cosas:
- Se trata de un gen muy conservado filogenéticamente.
- Ha sufrido una evolución muy rápida muy tardíamente. En la especie humana apenas hay variantes de este gen (polimorfismos) y las que hay son, como en el caso de la familia KE, muy negativas, no cabe duda de que nos hallamos ante un gen que ha sufrido una fuerte selección positiva en la especie humana.
Transmisión Autosómica Recesiva
En este tipo de transmisión sólo los homocigotos manifiestan el carácter y, por tanto, cada uno de sus progenitores debe tener en su genotipo al menos un alelo para ese locus. Los heterocigotos no manifiestan el rasgo, pero son portadores del alelo causante del mismo.
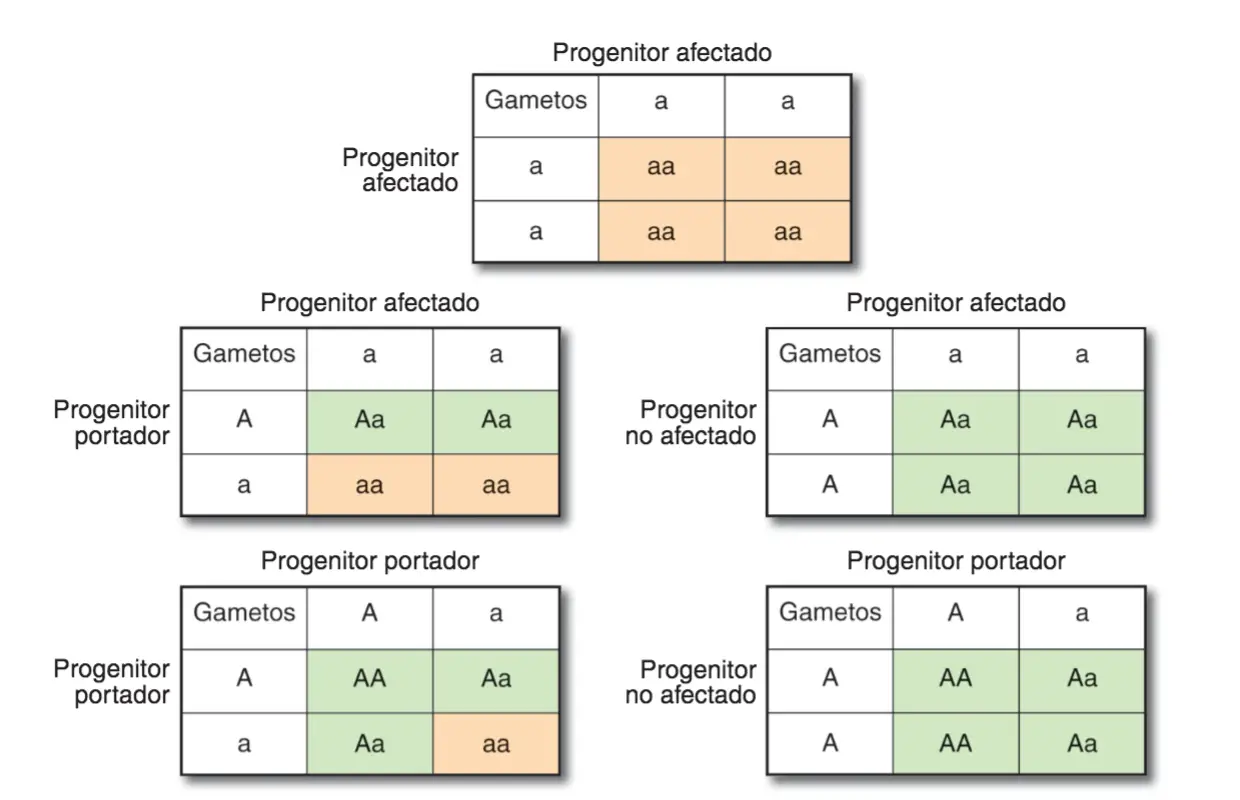
Descendencia esperada de diversos cruces, en relación a un carácter recesivo. En color crema se muestran los descendientes afectados y en verde los no afectados.
La Fenilcetonuria
La fenilcetonuria (PKU) es un ejemplo paradigmático de la relación entre genes y conducta, en este
caso, de herencia autosómica recesiva. Los sujetos que padecen la enfermedad tienen un cociente intelectuaI de alrededor 50, este es su fenotipo conductual.
Esta discapacidad cognitiva se debe a un alelo recesivo de un gen ubicado en el cromosoma 12 y su explicación está en la acumulación de fenilalanina porque no son capaces de metabolizar la fenilalanina a tirosina. Este se produce por alteración de producción de mielina, ocasionando en última instancia desmielenización de las fibras nerviosas cerebrales.
Transmisión Ligada al Sexo
A diferencia de los autosomas, de los que cada descendiente recibe una copia de cada progenitor, los cromosomas sexuales, X e Y, se heredan de una manera especial: el cromosoma Y, cuya presencia determina el sexo masculino en mamíferos, sólo se trasmite de macho a macho; por su parte, el cromosoma X de los machos siempre procede de la madre. Los machos son haploides (o hemicogóticos, al tener sólo una copia de cada gen del cromosoma X), es evidente que en ese caso no se puede hablar de dominancia o recesividad para los alelos de dichos genes.
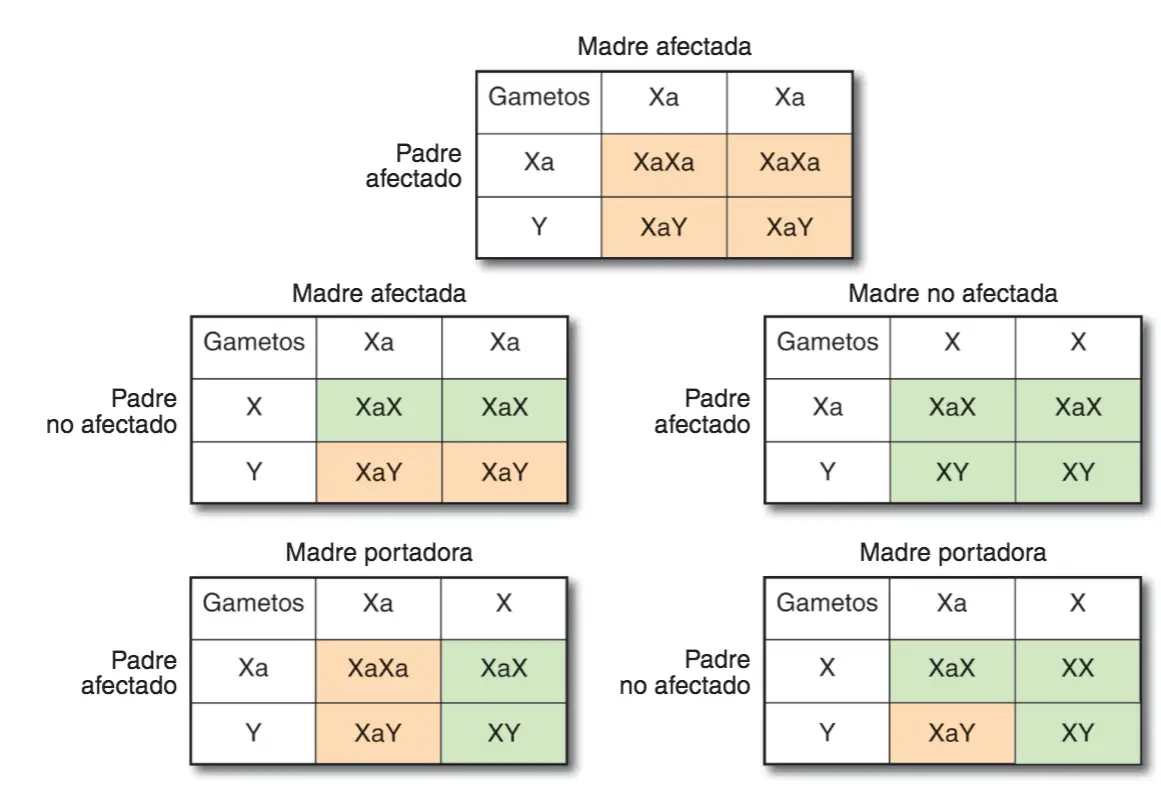
los descendientes afectados y en verde los no afectados.
En las hembras, la cuestión es más compleja porque uno de los dos cromosomas X está inactivado, pero sus tejidos son genéticamente mosaicos, en el sentido de que en unos grupos de células es el X recibido del padre el que está inactivado y en otros el X de la madre: el fenotipo de las células dependerá en cada caso del X activo.
Enfermedades ligadas al cromosoma X de carácter recesivo:
- Daltonismo. Incidencia 8% machos, y un 0,04% hembras. Consiste en la alteración producida por el alelo del daltonismo impide que la persona afectada por la enfermedad sea capaz de distinguir qué cifra aparece en el dibujo (forma parte del test de lshihara).
- Hemofilia A. El alelo responsable de la enfermedad causa una deficiencia en el factor VIII que impide que la sangre coagule normalmente.

El Síndrome de X Frágil
El llamado síndrome de X frágil es la segunda causa de discapacidad mental moderada en varones (1 de cada 1250 varones por 1 de cada 2500 mujeres) , sólo por detrás del síndrome de Down. Las mujeres portadores tienden a presentar una cierta sintomatología, si bien menos severa … Además de un CI moderadamente bajo, tres cuartas partes de los varones que presentan X frágil adoptan algunas conductas características: alteraciones de la expresión hablada (habla repetitiva y/o desorganizada), aversión a mantener la mirada, conducta autista …
Citológicamente, el síndrome de X frágil se manifiesta por una rotura o estrechamiento en la posición q2 7.3 del cromosoma X. La causa parece ser la mutación de un gen que se expresa en el tejido cerebral. Esta mutación consiste en un aumento espectacular en el número de repeticiones del triplete CGG: mientras que en sujetos normales este triplete está repetido entre 6 y 54 veces, en sujetos que padecen el síndrome el número de repeticiones es de más de 200 veces.
ANÁLISIS GENÉTICO DE LA CONDUCTA HUMANA
El Gen SRY y el Fenotipo Masculino
Hay un gen cuya relación directa con el fenotipo está bastante demostrada, el gen SRY (acrónimo en inglés de región determinante del sexo del cromosoma Y). Este gen se asocia indisolublemente con la masculinidad. El gen SRY se activa entre las 6ª y las 8ª semana de gestación, y podemos asegurar que se trata de un gen regulador porque codifica un factor de trascripción.
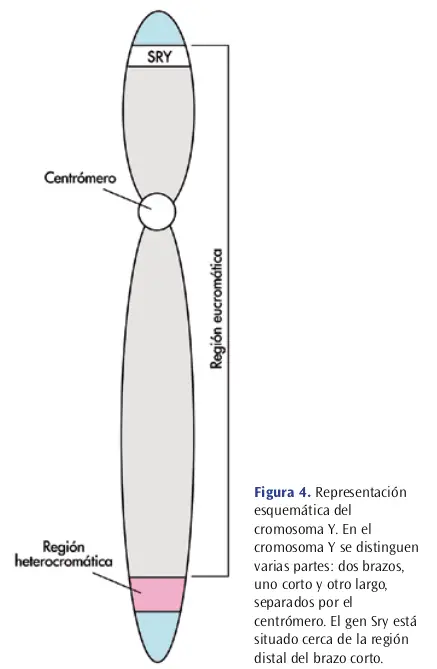
Seguramente, la proteína codificada por el gen SRY (el hipotético Factor Determinante de Testículos, o FDT) inicia una cascada de acontecimientos consistente en la expresión de otros muchos genes (destaca el gen SOX9).
- El gen SRY se halla presente en todos los varones XY.
- Los sujetos XY a los que por deleción les falta el locus de este gen SRY, son fenotípicamente mujeres.
- Lo mismo ocurre cuando dicho gen ha sufrido una mutación que lo hace incapaz de funcionar correctamente, bien por no sintetizarse la proteína codificada, bien porque la proteína resultante no es funcional.
- Cuando el gen SRY se encuentra por traslocación es sujetos cuyo cariotipo es 46,XX, el fenotipo es siempre masculino.
Sorprendentemente, en casos de sujetos XX (y ausencia de gen SRY) donde por duplicación existe una copia extra de este gen SOX9, el fenotipo es masculino: ello es debido a que, en último extremo, es la cantidad de producto sintetizado a partir del gen SOX9 lo que inclina la balanza hacia la diferenciación como testículos de las gónadas bipotenciales. La insuficiencia funcional del gen SOX9 da lugar, entre otras cosas, a fenotipo femenino en un gran porcentaje de casos (75%).
Genética y Epigenética de la Conducta: la Testosterona y la Diferenciación Sexual
Los genes SRY y SOX9 son necesarios pero no suficientes y lo demuestra la existencia del llamado Síndrome de Feminización Testicular (en Psicología Fisiológica «Síndrome de Insensibilidad a los andrógenos»). Cuando la insensibilidad a los andrógenos (que es la causa de este síndrome) es completa, la orientación sexual es típicamente femenina. Esta insensibilidad a los andrógenos se debe a una mutación del gen que codifica el receptor de andrógenos, lo que impide que la testosterona llegue al núcleo de las células y ejerza sus funciones regulatorias, cuyos efectos se manifiestan en la diferenciación sexual masculina, crecimiento muscular, desarrollo de los huesos, espermatogénesis y crecimiento de la próstata. Curiosamente, este gen se encuentra en el cromosoma X.
Otra mutación genética cuyos efectos son en cierta manera complementarios a los observados en el caso de la insensibilidad a los andrógenos es el síndrome de Hiperplasia Adrenal Congénita. Se trata de un rasgo autosómico recesivo consistente en un deficiencia en el gen CYP2 7 que impide que se sintetice la enzima 21-hidroxilasa esteroide adrenal. Las mujeres que portan esta mutación, nacen con unos genitales externos de apariencia masculina, si bien los genitales internos son típicamente femeninos.
Genética del Ritmo Circadiano
Prácticamente todos los animales, de insectos a humanos, exhiben de modo espontáneo ritmos circadianos (de alrededor de un día). No hay duda de que la adecuación de los procesos fisiológicos a este ritmo geológico es esencial para la adaptación biológica: así cabe explicar la existencia de mecanismos celulares endógenos que regulan la fisiología y la conducta de modo acorde con dicho ciclo geológico; son los llamados relojes biológicos. En los mamíferos, el mecanismo de este reloj circadiano endógeno se haya en el núcleo supraquiasmático del hipotálamo y funciona gracias a la acción coordinada de determinados genes.
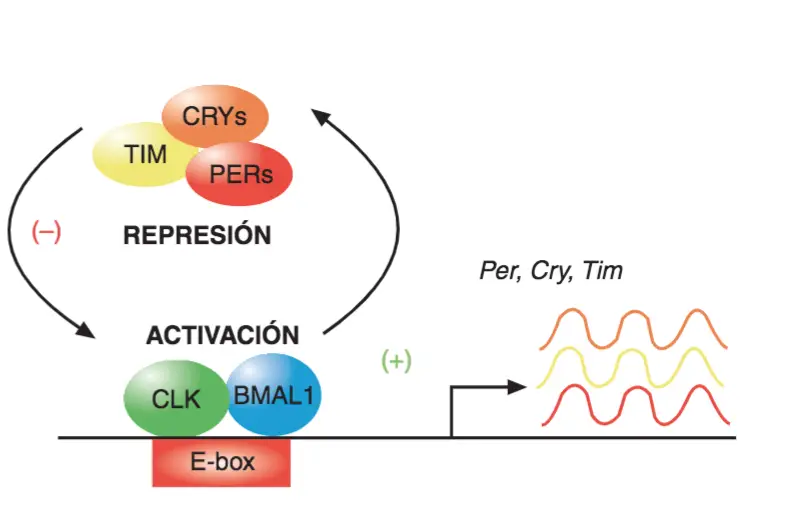
El modelo trata de representar los bucles de retroalimentación positiva y negativa vía regulación de la trascripción de proteínas reguladoras tal como se supone que tiene lugar en el núcleo supraquiasmático del hipotálamo de los mamíferos: las proteínas BMAL 1 Y CLK se unen al promotor de otros genes activando su trascripción, concretamente de los genes Per, Cry y Tim; Esta activación da lugar a la síntesis de los ARNm correspondientes (líneas oscilantes) y, cuando estos ARNm llegan al citoplasma dan lugar a la síntesis de las proteínas PER, CRY y TIM que eventualmente ingresan en el núcleo, tras sufrir modificaciones que hacen que interactúen entre sí y se estabilicen. Una vez dentro del núcleo entran en contacto con el complejo proteínico CLK-BMAL 1 justamente en la zona donde estas dos proteínas habían inducido la trascripción de los genes Per, Cry y Tim, lo que inhibe la trascripción de estos genes, cerrando el bucle de retroalimentación negativa de la regulación de los genes del reloj biológico. Al formarse el complejo CLK-BMAL 1 + PER-TIM-CRY, se va perdiendo la afinidad de la unión de las proteínas PER-TIMCRY, que acaban disociándose y, cuando esto ocurre, de nuevo el complejo CLK-BMAL 1 recupera su capacidad de activar la trascripción de los genes Per, Cry yTim. Las proteínas PER, TIM y CRY que se hallan en el núcleo, se degradan. El ciclo completo tiene una duración de 24 horas.
Se ha demostrado que existen variantes (alelos) de algunos de estos genes involucrados en la regulación del ritmo circadiano de los mamíferos, alelos que permiten explicar algunas diferencias fenotípicas en lo que se refiere al ciclo natural de actividad/ inactividad propio de los mamíferos:
- En hámster, se observó que en algunos casos el ciclo de actividad/inactividad era más corto de las 24 h. habituales; esta variante genética recibió el nombre de tau. Se demostró que los homocigotos para el gen tau presentaban un ciclo de 20 h., mientras que el de los heterocigotos era de 22 h.
- En humanos, el Síndrome de Fase Adelantada del Sueño (ASPS), guarda un estrecho paralelismo con la variante tau 15 anteriormente citada: los miembros afectados de familias donde este síndrome es frecuente, al parecer, sufren una alteración en el gen PER2. Estas personas se caracterizan por dormirse entre las 6 y las 9 de la tarde y despertarse entre las 2 y las 5 de la madrugada, mucho más pronto de lo habitual. La sustitución del aminoácido serina por el aminoácido glicina da lugar a que la proteína PER2 se acumule más deprisa, acelerando el bucle de feedback del reloj y, como consecuencia ocasionando un período circadiano más breve.
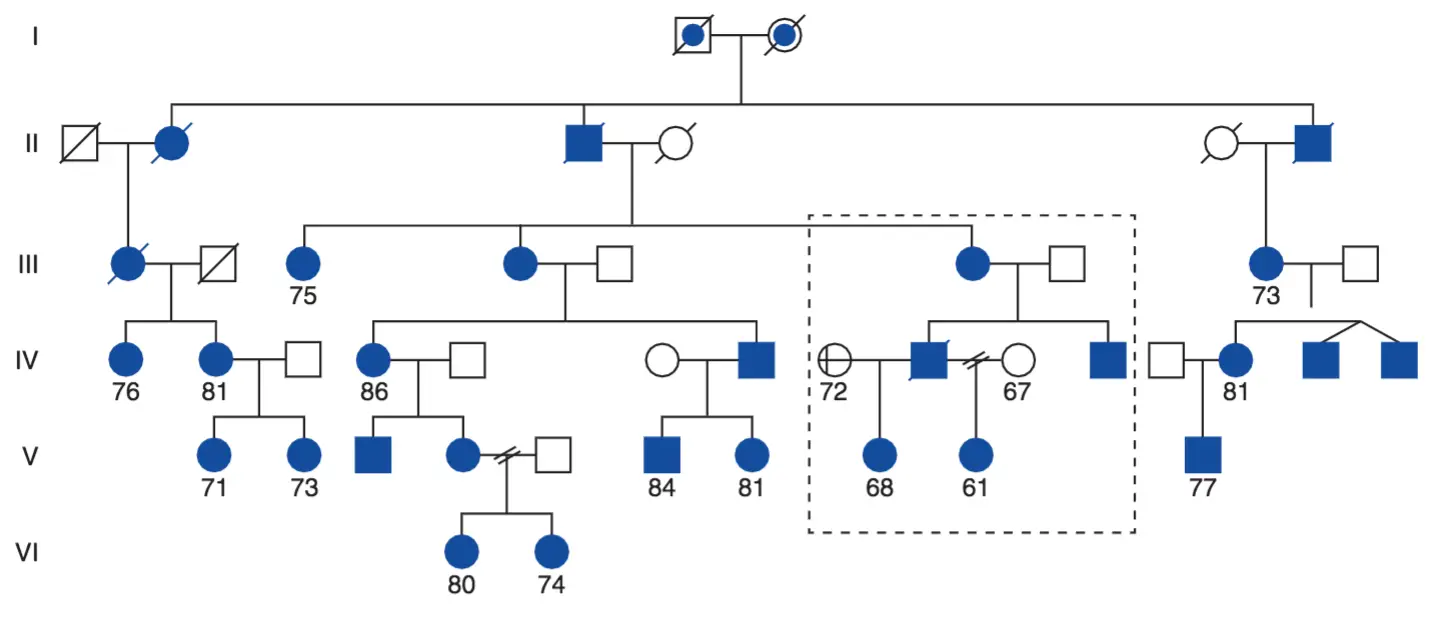
Pedigrí parcial de una familia extensa compatible con un fenotipo autosómico dominante: los sujetos afectados sufren un síndrome conocido como de fase avanzada del sueño (ASPS por sus siglas en inglés).
- El Síndrome de Fase Demorada del Sueño (DSPS) afecta a menos del 1 % de la población y se caracteriza justamente por la imposibilidad de dormirse o despertarse a voluntad a la hora deseada. Estas personas tienen que esperar hasta más tarde de las 3.00 h. para dormirse, y se despiertan entre las 10.00 y las 15.00 h. si pueden a placer. Hasta ahora sólo se ha asociado una variante del gen PER3 con el DSPS.
Hay otros síndromes que afectan al sueño, pero que no están relacionados con los ritmos circadianos.
- Insomnio Familiar Fatal. Se trata de una enfermedad autosómica dominante caracterizada, además del insomnio intratable, por trastornos motores, un deterioro cognitivo consistente en pérdida de la capacidad de atención, déficits en la memoria a corto plazo, y en general un proceso de degeneración de los procesos cognitivos que termina en demencia y muerte. El alelo que causa este síndrome se encuentra es una variante del mismo gen que produce el Síndrome de Creutzfeld-Jakob (gen PRNP de la proteína priónica) y se halla en el cromosoma 20.
La Narcolepsia
La Narcolepsia es un trastorno neurológico relacionado con el sueño. Consiste en una incapacidad para regular el patrón circadiano de sueño-vigilia normal. Los pacientes sufren además somnolencia diurna excesiva, parálisis del sueño y alucinaciones hipnagógicas así como, en la mayoría de los casos, cataplexia. La cataplexia aparece con mayor frecuencia en situaciones estresantes o intensamente emotivas. Es frecuente además, que estos pacientes entren directamente en la fase REM del sueño, En la fase de sueño REM el tono muscular es más bajo y, por tanto, la inmovilidad más completa: podría considerarse que la cataplexia es el correlato motor normal del sueño REM.
Uno de los avances más importantes en el conocimiento de las bases fisiológicas de esta enfermedad (afecta entre 0.03 y 0.1 % en la población general), proviene de la genética. Una mutación del gen que codifica el receptor de hipocretina (neurotrasmisor participa en la regulación de los niveles de alerta o vigilancia y también en la regulación de la ingesta).
Genética del Hambre y Obesidad
La obesidad humana puede explicarse por diferentes factores:
- El acceso ilimitado a alimentos de alto valor energéticos.
- Factores de riesgo asociados a genes concretos.
- Gen responsable de la producción de leptina. La leptina es una hormona peptídica producida por los adipocitos, que regula la ingesta actuando sobre receptores hipotalámicos. El efecto de la leptina se atribuye a que potencia la señal de saciedad que la ingesta de comida provoca, además de reducir su valor hedónico. Una deleción de guanina en la posición 133 que da lugar a una leptina incompleta y, en este caso, fisiológicamente inactiva.
- Gen que codifica el receptor neuronal de leptina. Su mutación determina un truncamiento de la proteína receptora que anula la funcionalidad de la leptina.
- Gen que codifica el receptor de melanocortina MC4R. Su mutación produce una pérdida total de función de este receptor de melanocortina MC4R ocasionan en los sujetos portadores hiperfagia y obesidad. Los sujetos homocigóticos para el alelo mutante alcanzan un grado todavía mayor de obesidad.
Genética, Neurotransmisores y Conducta Humana
El exceso o carencia de cada uno de los neurotransmisores suele relacionarse con alteraciones muy notables de la conducta humana. La acción de los neurotransmisores se ejerce en un contexto anatómico-funcional sumamente importante, la sinapsis. En estrecha relación con los neurotransmisores están las proteínas receptoras (los receptores ), de las que depende todo el efecto que los neurotransmisores puedan llegar a tener sobre la fisiología neuronal. Las enzimas que catalizan la síntesis y degradación de los neurotransmisores, así como las proteínas trasportadoras, pueden tener una influencia muy notable en lo que se refiere a la concentración sináptica de dichos neurotransmisores tanto a corto como a largo plazo. Los neurotransmisores, serotonina y dopamina, son los que, de momento, han demostrado más relación con algunas conductas humanas.
La Serotonina (5-HT)
La serotonina es un neurotransmisor producido sobre todo por neuronas de los núcleos del rafe, y su función parece ser la de modular la actividad fisiológica de las neuronas sobre las que actúa. Es un hecho de que sujetos que padecen depresión presentan niveles bajos de serotonina, así como que se ha asociado una escasez de este neurotransmisor con dificultades para el autocontrol y un despliegue excesivo de conductas agresivas.
Uno de los genes más estudiados es el que codifica el transportador de serotonina (SERT, por sus siglas en inglés), del cual existen en la población humana dos alelos, uno largo (I) y otro corto (s).
- Homocigóticos para el alelo s. tienden a presentar con mayor frecuencia ansiedad y tendencia a evitar las situaciones amenazantes, así como a puntuar más alto en el rasgo «neuroticismo ». La función del transportador de serotonina (SERT) es devolver este neurotransmisor a la neurona presináptica desde el espacio sináptico, lo que constituye una forma de inactivación del neurotransmisor.
- Cuando la serotonina (o cualquier neurotransmisor) permanece en el espacio sináptico, aumenta la probabilidad de que entre en contacto con los receptores postsinápticos y se active la neurona postsináptica. Al sufrir un bloqueo constante de la recaptación durante todo el desarrollo, los procesos de regulación en los receptores hacen que la actividad serotoninérgica sea menor.
- Los homocigóticos para el alelo s, pero también, aunque en menor medida los heterocigóticos (Is), se ven muy afectados por experiencias traumáticas durante el desarrollo (estrés, maltrato, abusos, etc.) en el sentido de que estas situaciones provocan con mayor frecuencia trastornos depresivos que en los homocigóticos para el alelo I.
- Los individuos portadores del alelo s muestran una hiperreactividad de la amígdala, una estructura cerebral especialmente involucrada en el procesamiento emocional. La posible explicación de estos efectos podría estar en que los niveles elevados de serotonina durante el desarrollo deterioran de forma permanente los circuitos reguladores que conectan el rafe dorsal (estructura troncoencefálica cuyas neuronas liberan serotonina) con la corteza prefrontal y la amígdala.
Otro gen involucrado en la regulación de los niveles de serotonina (y también de noradrenalina) es el gen de la monoaminoxidasa A (MAOA). Se ha demostrado que este gen era inactivo (o poco activo) correlaciona con la impulsividad y conductas violentas y/o antisociales.
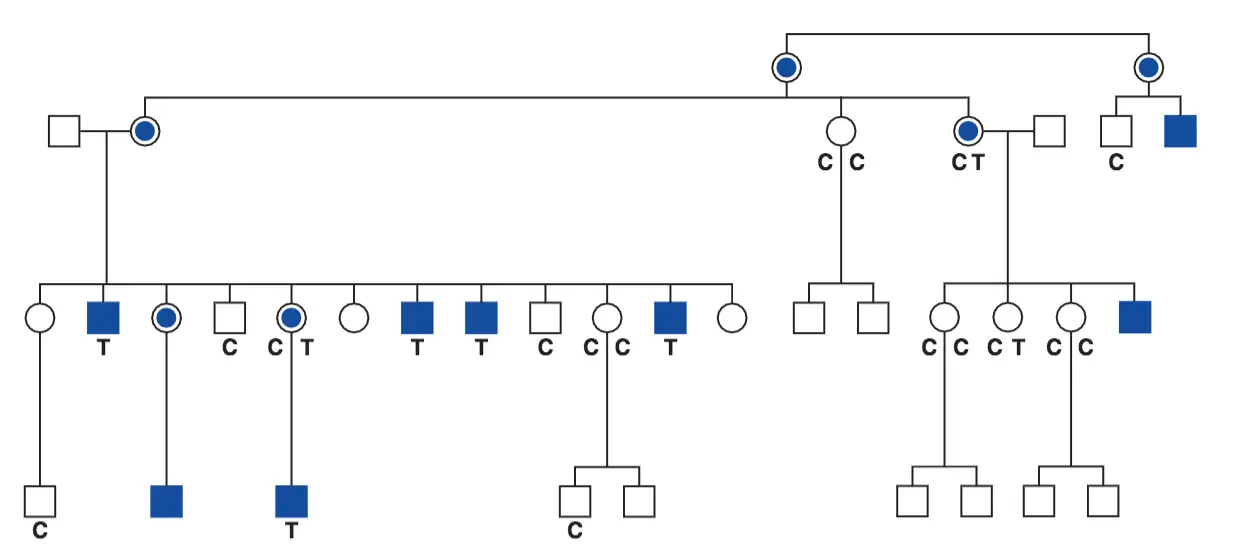
Pedigrí de una familia holandesa alguno de cuyos miembros se vieron afectados de una cierta discapacidad mental acompañada de prominentes trastornos conductuales, entre ellos agresión impulsiva, intento de violación, exhibicionismo … Los 5 varones bioquímicamente analizados cuyo fenotipo era afectado, presentaban un deficiencia completa y específica en la actividad de la enzima monoaminooxidasa A (MAOA), debida a una mutación puntual en el nucleótido 936, donde la Citosina (C) habías sido sustituida por Timina (T). Las mujeres portadoras analizadas fueron heterocigóticas CT, mientras que los varones analizados no afectados presentaban siempre la variante alélica C.
Dopamina
- Variaciones en los niveles de actividad de la dopamina se han relacionado con:
- Alteraciones motoras propias del Parkinson, asociadas a bajos niveles de dopamina.
- Deterioro conductual propio de la esquizofrenia asociado a niveles elevados de dopamina.
- Se ha comprobado con ratones knockout a los que se les ha inactivado el gen que codifica la proteína responsable de la extracción de la dopamina del espacio sináptico, que la actividad motora espontánea está modulada por este neurotrasmisor.
- Por otra parte, se ha detectado en humanos un polimorfismo en la proteína que constituye el receptor D4 de dopamina: los individuos que portan una variante determinada, concretamente la codificada por el alelo llamado largo, tienden a ser personas ávidas de novedad y buscadores de situaciones placenteras, en mucha mayor medida que quienes sólo portan la variante corta del gen.
- El receptor D4 se expresa en el hipotálamo y la parte del sistema límbico más especialmente involucrada en procesos emocionales, así como en la corteza prefrontal, y parece involucrado en los procesos de atención y otras funciones cognitivas superiores.
- El hecho de que el receptor D4 ejerza funciones inhibitorias sobre las neuronas, especialmente las del córtex prefrontal, unido al hecho de que la variante larga sea menos eficaz, hace verosímil la relación causal entre esta variante y algunas alteraciones observadas en el comportamiento de diferentes individuos, especialmente el síndrome de déficit de atención e hiperactividad y también con el rasgo de personalidad tipificado como “buscador de sensaciones”. Los portadores de este alelo puedan ser propensos a hacerse adictos a sustancias de abuso.
- Tanto el transportador de dopamina DAT1 como el receptor DR4D se han asociado con el trastorno de déficit de atención con hiperactividad (TDAH):
- Los homocigóticos para la variante larga (10 repeticiones) de DAT1 presentan más hiperactividad.
- Los homocigóticos para la variante larga del receptor DR4D (7 repeticiones) muestran sobre todo síntomas de déficit de atención.
- Es probable que ambos síntomas estén relacionados con alteraciones funcionales en diferentes zonas cerebrales y hayan de ser abordados de modo específico.
ALTERACIONES CROMOSÓMICAS Y CONDUCTA
Síndrome de Williams
El síndrome de Williams se debe a una deleción de un segmento del cromosoma 7, concretamente la sección 7q11.2 .Estos pacientes son hemicigóticos para los genes correspondientes, sólo tienen una copia de las dos que corresponden a los individuos diploides normales.
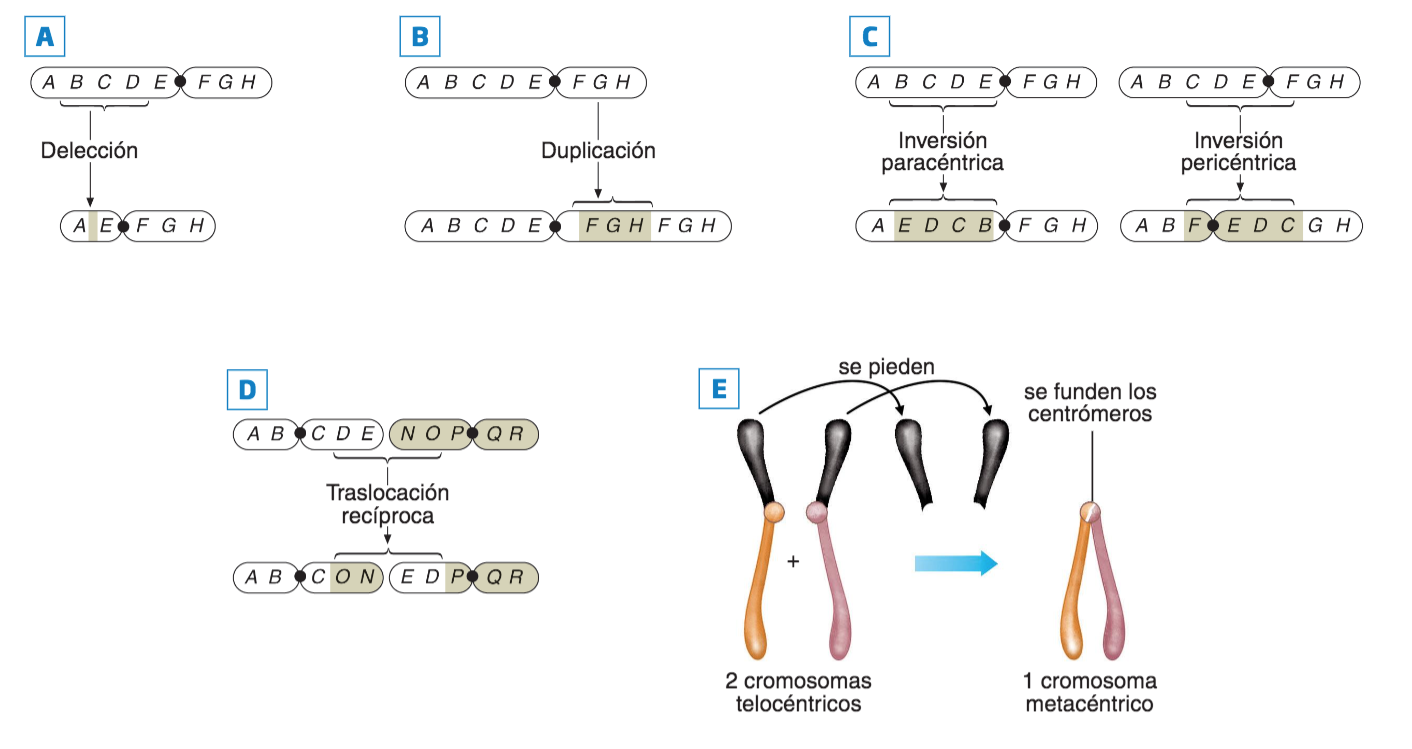
Alteraciones cromosómicas estructurales y análisis de su morfología mediante técnicas de bandeo. A. Deleción. B. Duplicación. C. Inversión. D. Traslocación Recíproca. E. Traslocación Robertsoniana.
En la deleción se pierden hasta 20 genes, y cuanto más grande es la deleción, más severos son los síntomas.
Rasgos característicos.
- CI bajo (alrededor de 50).
- Notable incapacidad para el aprendizaje espacial y numérico, para la resolución de problemas y para la planificación, así como para dibujar y escribir.
- Notable capacidad lingüística, muy superior a lo esperable por su CI, tanto en lo que se refiere a la sintaxis como al vocabulario.
- Muy comunicativos y confiados cuando conocen gente nueva, tienen facilidad para reconocer rostros.
- Algunos están especialmente dotados para la música.
Impresión Genómica y Epigenética de la
Conducta
La manifestación fenotípica de un alelo depende de que sea dominante o recesivo, además, el efecto de la expresión de un gen es predecible según el genotipo, homocigótico o heterocigótico. Además, sabemos que lo normal es que ambos alelos, si son funcionales, se expresen por igual (expresión bialélica). Sin embargo, a finales de la década de 1970 se demostró que la forma de expresarse de algunos genes no se ajusta a lo establecido, sino que difiere en función únicamente de si el alelo procede del padre o de la madre: es lo que se llama impresión o grabación genómica.
En la especie humana se dan dos síndromes de origen genético, el de Prader-Willi y el de Angelman, que demuestran que existe impresión gamética (sinónimo de impresión genómica). Ambos síndromes aparecen cuando se da una deleción en el brazo largo del cromosoma 15.
Síndrome de Prader-Willi
- Se produce cuando el cromosoma que ha sufrido la deleción es el del gameto masculino.
- Obesidad.
- Apetito desmedido.
- Discapacidad mental.
En el síndrome de Prader-Will sólo están presentes los genes maternos correspondientes, (los del padre se perdieron en la deleción). Dado que la impronta genómica consiste en la inactivación de genes en función del sexo del productor de los gametos, esto significa que estos sujetos es como si carecieran por completo de algunos genes, los que faltan por deleción y los que no se expresan por improntación en el gameto femenino.
Síndrome de Angelman
- Se produce cuando el cromosoma que ha sufrido la deleción es el del gameto femenino.
- Discapacidad mental grave.
- Risa convulsiva.
- Movimientos involuntarios (tipo marionetas).
En el síndrome de Angelman sólo están presentes los genes paternos correspondientes, (los de la madre se perdieron en la deleción). Dado que la impronta genómica consiste en la inactivación de genes en función del sexo del productor de los gametos, esto significa que estos sujetos es como si carecieran por completo de algunos genes, los que faltan por deleción y los que no se expresan por improntación en el gameto masculino.
Cuando no hay deleción, pero se han heredado de la madre las dos copias del cromosoma 15 (disomía uniparental), también aparece el síndrome de Prader-Willi (puesto que el alelo correspondiente de ambos cromosomas está improntado/inactivado). Cuando ambos cromosomas proceden del padre, aparece el síndrome de Angelman.
En determinados embarazos humanos, ocurre que un espermatozoide fecunda un óvulo que carece de núcleo; se conoce con el nombre de mola, una masa placentaria sin presencia de feto: el análisis genético demuestra que todo el genoma procede del espermatozoide (en este caso parece ser que los cromosomas del espermatozoide se duplican). El hecho de que no haya aportación materna ocasiona un desarrollo enormemente anómalo; se trata, pues, de un ejemplo de impresión genómica.
Síndrome de Down
El Síndrome de Down fue descrito por John Langdon Down en 1866. A finales de los años 50 del siglo XX, se descubrió que la presencia de un cromosoma 21 extra (trisomía) es la causa del Síndrome de Down. La explicación es una no disyunción meiótica.
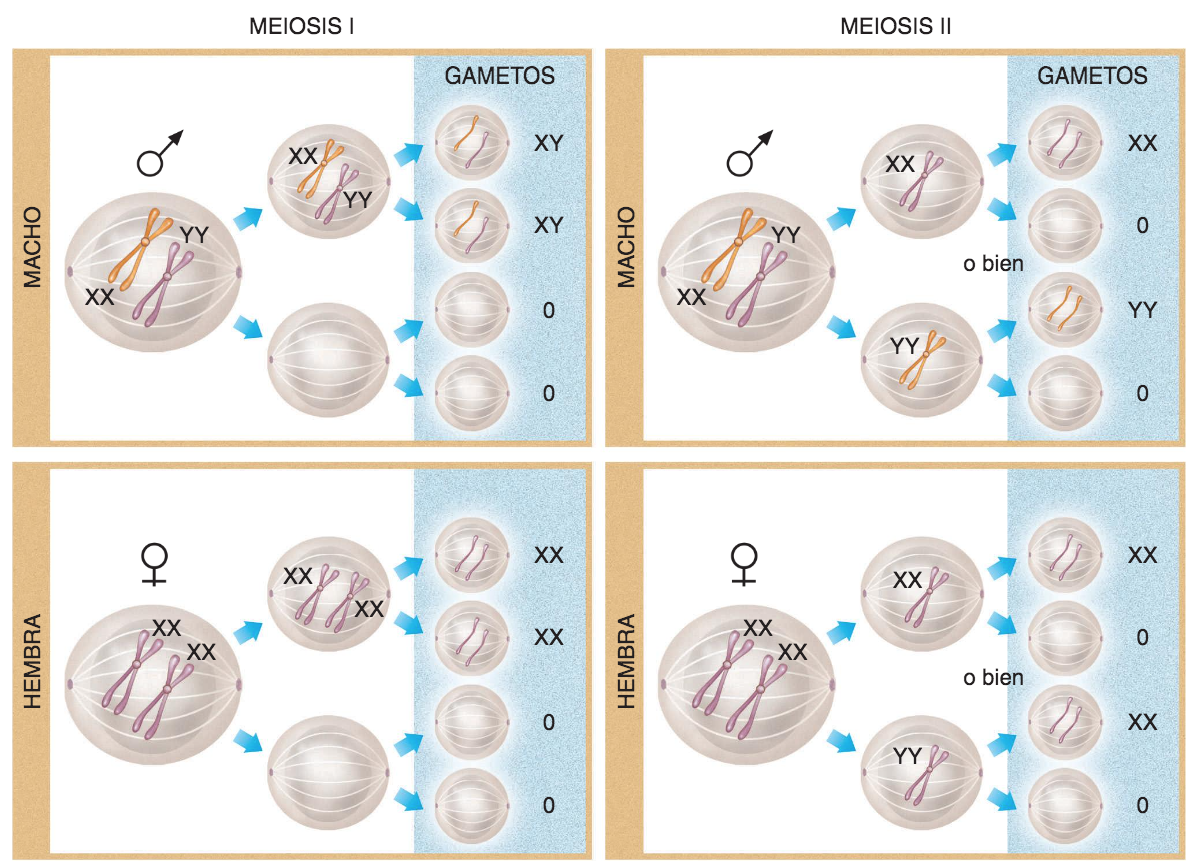
La incidencia es de 1 de cada 800 nacidos vivos. El factor de riesgo más importante del síndrome de Down es la edad de la madre. Una posible explicación del efecto de la edad sobre las trisomías puede ser el que a medida que la mujer se hace mayor, pierde eficacia el proceso por el cual se abortan los embriones cromosómicamente anormales: es lo que se conoce como selección materna.
Ocasionada la trisomía esta causada por la no disyunción en la gametogénesis femenina (formación del óvulo), sin embargo, en un 5-6 % de los casos, la trisomía es de origen paterno. Existe una variante del síndrome de Down llamada familiar, que implica una traslocación robertsonina. Igualmente, cabe la posibilidad de que una duplicación de segmentos del cromosoma 21 pueda dar lugar a un fenotipo similar al que presentan los sujetos Down.
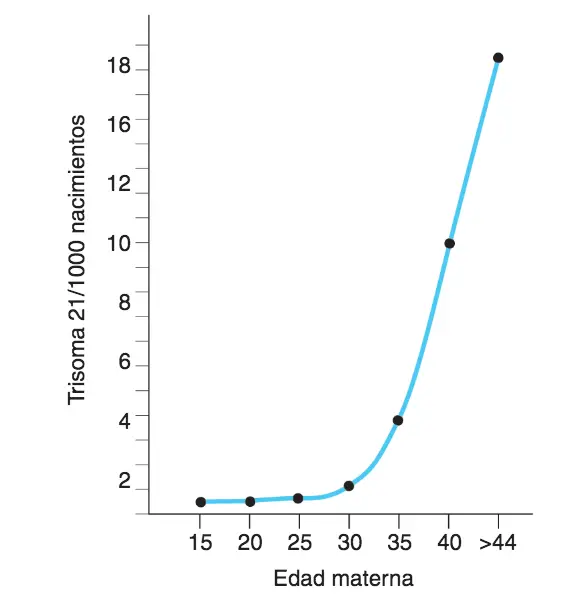
Representación gráfica de la relación entre la edad de la madre y la incidencia del Síndrome de Down por trisomía del par 21.
Síndrome de Down
- La causa del Síndrome de Down es la presencia de un cromosoma 21 extra (trisomía).
- Baja inteligencia: su CI promedio es 55.
- Portan tres copias del gen de la proteína precursora de amiloide (APP).
- Reducción en el tamaño de la corteza prefrontal.
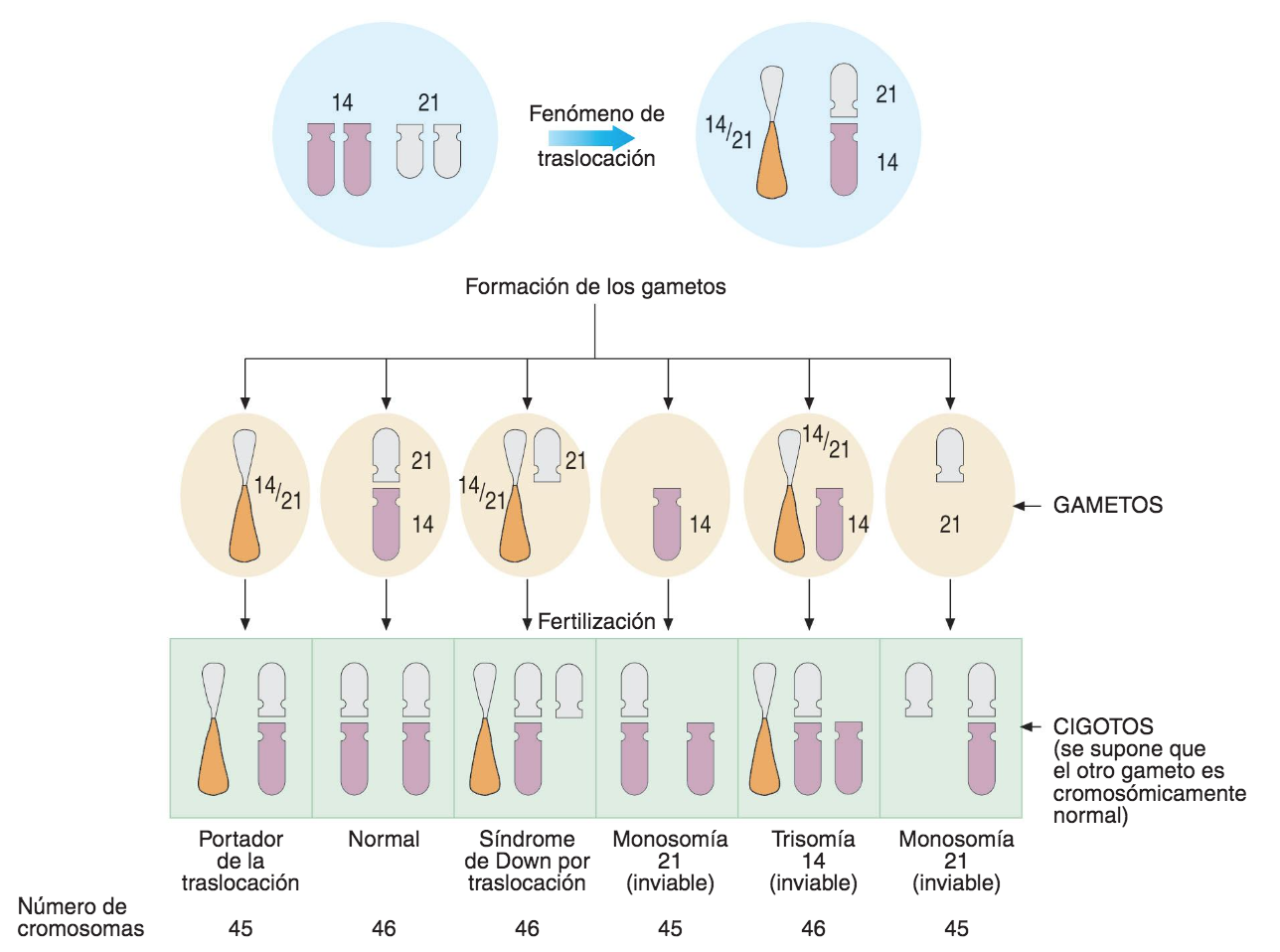
Explicación citológica del Síndrome de Down familiar o atribuible a traslocación robertsoniana 14/21. Cuando un sujeto normal fenotípicamente hablando es portador de una traslocación robertsonina 14/21 , puede producir diversos tipos de gametos en función de cómo se repartan los cromosomas en la meiosis lo que, en algún caso, puede dar lugar a síndrome de Down.
Cromosomas Sexuales
El tipo de aneuploidía más frecuente en la especie humana es la trisomía. De entre ellas, las de los cromosomas sexuales y la del cromosoma 21 son las más compatibles con la vida, en tanto en cuanto son las más frecuentes en individuos vivos. La única monosomía compatible con la vida es la del cromosoma X: el llamado síndrome de Turner (45, X).
Síndrome de Turner
Es la única monosomía que se conoce compatible con la vida humana. Entre las características físicas más notables que presentan estos pacientes:
- Infertilidad.
- Ausencia de desarrollo de los rasgos sexuales secundarios, desarrollo insuficiente de los ovarios y vagina y útero pequeños e inmaduros.
- Estos pacientes son de baja estatura.
- Inteligencia verbal prácticamente normal.
- Inteligencia espacial y numérica por debajo de la media
- Problemas para reconocer las emociones, así como para relacionarse socialmente.
Cromosomas Sexuales, Sexo y Cromatina de Barr
¿Cómo es posible que en el síndrome de Turner (45,X), aparezcan deficiencias fenotípicas notables y, sin embargo, los varones posean un solo cromosoma X y sean normales en cuanto varones?. En las hembras de los mamíferos la mayor parte del 2° cromosoma X parece ser que se inactiva a partir de cierto momento, dando lugar a la llamada cromatina o corpúsculo de Barr. Esta cromatina de Barr siempre aparece cuando hay más de un cromosoma X y en un número exactamente igual al número de cromosomas X menos 1 (nX-1 ), independientemente del sexo fenotípico, no es ni más ni menos que
el cromosoma X inactivado (llamada hipótesis de Lyon). La inactivación de uno de los dos cromosomas X explica varias cosas:

- Si se expresaran todos los genes de ambos cromosomas X, las mujeres tendrían casi el doble de productos génicos de los genes ubicados en el cromosoma X.
- La existencia de tejidos en mosaico en las hembras. El mosaicismo solo se da en las hembras.
- La expresión de un gen llamado XIST que produce un ARN que, en vez de salir al citoplasma, se une al cromosoma X impidiendo su trascripción. La inactivación de uno u otro cromosoma X parece distribuirse al azar. Esta inactivación permite asegurar que la dotación génica de machos y hembras sea la misma.
- Hasta un 15% del cromosoma X inactivado escapa a dicha inactivación. Y precisamente uno de los genes que escapa a dicha inactivación es el causante (su falta) de la baja estatura de las mujeres Turner: es el gen SHOX .
Trisomía de los Cromosomas Sexuales
Hay tres trisomías de los cromosomas sexuales relativamente frecuentes que van acompañadas de fenotipos prácticamente normales. Estas anomalías cromosómicas son el resultado de un cigoto donde uno de los gametos que lo formaron habría sufrido una no disyunción meiótica.
- 47,XXX. Fenotipo femenino, prácticamente normal, estatura superior a la media, algunos casos de infertilidad, en muchos casos la tricotomía pasa desapercibida.
- 47,XXY (Síndrome de Klinefelter) fenotipo masculino. Son estériles, pueden presentar rasgos femeninos como como ginecomastia, problemas de conducta como inhibición social, déficit de atención con hiperactividad, media de CI de alrededor de 90, dificultades en el desarrollo de lenguaje.
- 47,XYY. Fenotipo masculino, pasa prácticamente desapercibida, más altos que la media, probable que tengan dificultades para el aprendizaje, especialmente del tipo verbal, así como hiperactividad e impulsividad.
AUTOEVALUACIÓN
REFERENCIAS
- Abril Alonso, A. (2016). Fundamentos de psicobiología (UNED (Sanz yTorres). Alcorcón (Madrid): Sanz y Torres.
- Powerpoint María Penado
- Apuntes Karla Marmolejo
- Apuntes Alejandra Mendieta
- YouTube

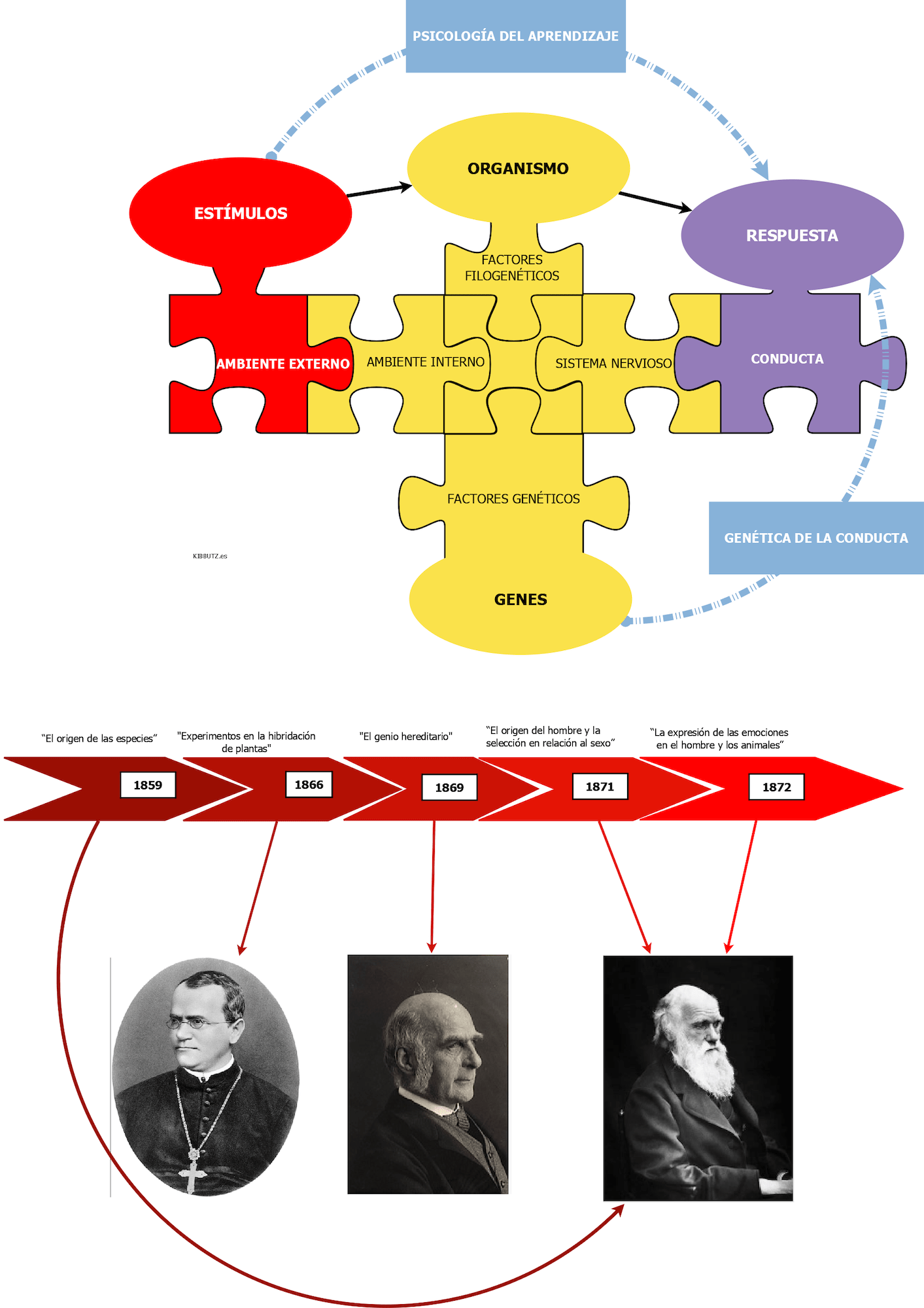{kind=link}